Please wait while the formulary information is being retrieved.
DRUG IMAGES
 BANOPHEN 25 MG TABLET
BANOPHEN 25 MG TABLET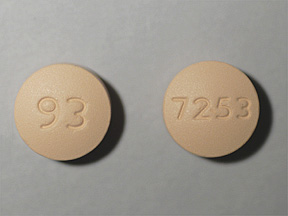 WAL-FEX ALLERGY 180 MG TABLET
WAL-FEX ALLERGY 180 MG TABLET ALLERGY RELIEF 10 MG TABLET
ALLERGY RELIEF 10 MG TABLET EQ ALLERGY RELIEF 180 MG TAB
EQ ALLERGY RELIEF 180 MG TAB EQ ALLERGY (LORAT) 10 MG TAB
EQ ALLERGY (LORAT) 10 MG TAB
The following indications for ALLERGY RELIEF (cetirizine hcl) have been approved by the FDA:
Indications:
Allergic conjunctivitis
Allergic reaction
Allergic rhinitis
Anaphylaxis
Chronic idiopathic urticaria
Cough
Dermatographic urticaria
Idiopathic parkinsonism
Insomnia
Motion sickness
Nasal congestion
Nausea and vomiting
Nausea
Parkinsonism
Perennial allergic rhinitis
Pruritus of skin
Seasonal allergic rhinitis
Sneezing
Urticaria
Vertigo
Vomiting
Professional Synonyms:
Agrypnia
Ahypnia
Allergy eye itch
Anaphylactic reaction
Atopic conjunctivitis
Atopic rhinitis
Autographism
Cnidosis
Dermatography
Dermographia
Dermographism
Dermography
Ebbecke's reaction
Emesis
Factitious urticaria
Intermittent allergic rhinitis
Itching wheals
Itchy eyes due to allergies
Itchy skin eruption
Nasal stuffiness
Nettle rash
Non-seasonal allergic rhinitis
Ocular itching due to allergies
Paralysis agitans
Periodic runny nose
Primary Parkinson's disease
Pruritic dermatitis
Queasy
Riders' vertigo
Seasonal allergy
Severe type I hypersensitivity reaction
Skin writing
Trembling palsy
Uredo
Urticaria factitia
Urticarial rash
Urtication
Vomit
Weal
Indications:
Allergic conjunctivitis
Allergic reaction
Allergic rhinitis
Anaphylaxis
Chronic idiopathic urticaria
Cough
Dermatographic urticaria
Idiopathic parkinsonism
Insomnia
Motion sickness
Nasal congestion
Nausea and vomiting
Nausea
Parkinsonism
Perennial allergic rhinitis
Pruritus of skin
Seasonal allergic rhinitis
Sneezing
Urticaria
Vertigo
Vomiting
Professional Synonyms:
Agrypnia
Ahypnia
Allergy eye itch
Anaphylactic reaction
Atopic conjunctivitis
Atopic rhinitis
Autographism
Cnidosis
Dermatography
Dermographia
Dermographism
Dermography
Ebbecke's reaction
Emesis
Factitious urticaria
Intermittent allergic rhinitis
Itching wheals
Itchy eyes due to allergies
Itchy skin eruption
Nasal stuffiness
Nettle rash
Non-seasonal allergic rhinitis
Ocular itching due to allergies
Paralysis agitans
Periodic runny nose
Primary Parkinson's disease
Pruritic dermatitis
Queasy
Riders' vertigo
Seasonal allergy
Severe type I hypersensitivity reaction
Skin writing
Trembling palsy
Uredo
Urticaria factitia
Urticarial rash
Urtication
Vomit
Weal
The following dosing information is available for ALLERGY RELIEF (cetirizine hcl):
Dosage should be individualized according to the patient's response and tolerance.
The nasal inhaler delivers about 50 mcg of fluticasone propionate per The usual adult oral dosage of diphenhydramine hydrochloride is 25-50 mg 3 metered spray. The commercially available preparation delivers about 120 or 4 times daily at 4- to 6-hour intervals, not to exceed 300 mg in 24 metered sprays per 16-g bottle; the container should be discarded after the hours. labeled number of actuations have been used since the correct dose per
inhalation cannot be assured if used for additional doses. Dosage of The usual adult IM or IV dose of diphenhydramine hydrochloride is 10-50 mg; intranasal fluticasone propionate should be adjusted according to in a few patients, up to 100 mg may be required. Some experts recommend a individual requirements and response; the lowest effective dosage should be dose of 25-50 mg.
The rate of IV administration should not exceed 25 used in order to minimize potential systemic effects of the drug. (See mg/minute. Cautions: Hypothalamic-Pituitary-Adrenal (HPA) Axis Suppression.) The
maximum daily dosage of intranasal fluticasone propionate should not exceed The maximum adult IM or IV dosage of diphenhydramine hydrochloride is 400 100 mcg in each nostril (total of 200 mcg daily). mg daily.
In patients with chronic renal impairment (creatinine clearance of 30 mL/minute or less), both oral bioavailability and peak plasma concentrations of loratadine and desloratadine may be increased compared with individuals with normal renal function. However, elimination half-lives of the drug and its active metabolite appear to be similar to those of individuals with normal renal function. Patients with renal impairment receiving loratadine for self-medication should be advised to consult a clinician before initiating therapy, since a different dosage may be recommended.
Therapy with loratadine conventional or orally disintegrating tablets or oral solution should be initiated at a dosage of 10 mg every other day in adults and children 6 years of age and older with a glomerular filtration rate less than 30 mL/minute and at a dosage of 5 mg every other day in children 2-5 years of age with renal insufficiency. In addition, therapy with the commercially available tablets containing loratadine in fixed combination with pseudoephedrine sulfate should be initiated in adults and children 12 years of age and older with a glomerular filtration rate less than 30 mL/minute at a dosage of 5 mg once daily when the 12-hour formulation is used or at a dosage of 10 mg every other day when the 24-hour formulation is used, since clearance of both loratadine and pseudoephedrine are decreased in such patients. Hemodialysis does not appear to affect the pharmacokinetics of loratadine or desloratadine.
The pharmacokinetics of loratadine and its active metabolite also may be altered in patients with hepatic impairment and dosage adjustment may be necessary. Therefore, patients with hepatic impairment receiving loratadine for self-medication should be advised to consult a clinician before initiating therapy, since a different dosage may be recommended. Therapy with loratadine conventional or orally disintegrating tablets or oral solution should be initiated at a dosage of 10 mg every other day in adults and children 6 years of age and older with hepatic failure and at a dosage of 5 mg every other day in children 2-5 years of age with hepatic failure.
Since fixed-ratio combination preparations do not permit individual titration of dosages, and clearance of loratadine is decreased more substantially than that of pseudoephedrine sulfate in patients with hepatic impairment, the manufacturer recommends that tablets containing loratadine in fixed combination with pseudoephedrine sulfate generally not be used in such patients.
Adjustment of fexofenadine hydrochloride dosage may be necessary in patients with renal impairment. Peak plasma fexofenadine concentrations increased by 87 or 111%, and elimination half-life increased by 59 or 72% in patients with mild (e.g., creatinine clearance of 41-80 mL/minute) or severe (creatinine clearance of 11-40 mL/minute) renal impairment, respectively, when compared with those observed in healthy individuals. In addition, peak plasma fexofenadine concentration increased by 82% and elimination half-life increased by 31% in those on hemodialysis (creatinine clearance of 10 mL/minute or less) compared with healthy individuals.
The manufacturer states that adults and children 12 years of age and older with impaired renal function or those on hemodialysis should receive an initial fexofenadine hydrochloride dosage of 60 mg daily (either given alone or in fixed combination with 120 mg of pseudoephedrine hydrochloride (Allegra-D(R) 12 Hour)). The fixed-combination preparation containing 180 mg of fexofenadine hydrochloride and 240 mg of pseudoephedrine hydrochloride (Allegra-D(R) 24 Hour) generally should be avoided in patients with renal impairment because of a possible risk of accumulation of pseudoephedrine.
Children 6 to younger than 12 years of age with impaired renal function should receive an initial fexofenadine hydrochloride dosage of 30 mg daily.
Since the pharmacokinetics of fexofenadine do not appear to be altered in patients with hepatic impairment, the manufacturer states that dosage adjustment is not necessary in such patients. The manufacturer of Allegra-D(R) 12 Hour and Allegra-D(R) 24 Hour does not make specific recommendations for dosage adjustment in patients with hepatic impairment, although it is not known if pharmacokinetics of pseudoephedrine are altered in patients with hepatic impairment.
The manufacturer states that patients 12 years of age or older who have impaired renal function (e.g., creatinine clearance of 11-31 mL/minute) or hepatic impairment or who are undergoing hemodialysis (creatinine clearance of less than 7 mL/minute), should receive a cetirizine hydrochloride dosage of 5 mg daily. The manufacturer also states that children 6-11 years of age with impaired renal or hepatic function should use the lower recommended dosage (5 mg once daily). The manufacturer states that use of cetirizine hydrochloride in children younger than 6 years of age with impaired renal or hepatic function is not recommended because administration of doses smaller than 2.5
mg is difficult and not reliable, and pharmacokinetic data are lacking in this patient population.
When extended-release tablets of cetirizine hydrochloride in fixed combination with pseudoephedrine hydrochloride are used in patients 12 years of age or older who have impaired renal function (i.e., creatinine clearance of 11-31 mL/minute) or hepatic impairment or who are undergoing hemodialysis (creatinine clearance of less than 7 mL/minute), the recommended cetirizine hydrochloride dosage is 5 mg once daily.
When diphenhydramine was available only by prescription, the prescribing information for the drug indicated a usual oral diphenhydramine hydrochloride dosage for children weighing more than 9.1 kg of 12.5-25 mg 3 or 4 times daily at 4- to 6-hour intervals and for children weighing 9.1
kg or less an oral diphenhydramine hydrochloride dosage of 6.25-12.5 mg 3 or 4 times daily at 4- to 6-hour intervals.
However, these dosage recommendations are not included in the current labeling of nonprescription oral diphenhydramine preparations, and clinicians should use caution when considering use of nonprescription oral diphenhydramine in children younger than 4 years of age. (See Cautions: Pediatric Precautions.)
Alternatively, for oral, deep IM, or IV therapy, children (other than premature or full-term neonates) may be given 5 mg/kg daily or 150 mg/m2 daily divided in 4 doses; some experts recommend a dosage of 1-2 mg/kg daily. The rate of IV administration should not exceed 25 mg/minute.
The maximum oral, IM, or IV dosage of diphenhydramine hydrochloride in children older than 1 month of age is 300 mg daily.
For temporary relief of pruritus and pain associated with various skin conditions in adults and children 2 years of age or older, creams, lotions, or solutions containing 1-2% diphenhydramine hydrochloride are applied to the affected areas 3 or 4 times daily or as directed by a clinician; topical diphenhydramine should not be used more often than directed.
If the condition worsens, or if symptoms persist for longer than 7 days or resolve and then recur within a few days, topical therapy with diphenhydramine hydrochloride should be discontinued and a clinician consulted; the possibility of sensitization by, or hypersensitivity to, the drug should be considered.
Topical preparations containing diphenhydramine hydrochloride should not be used on large areas of the body or concomitantly with other preparations containing the antihistamine, including those used orally, since increased serum concentrations of diphenhydramine may occur that can result in systemic toxicity. (See Acute Toxicity: Manifestations, in the Antihistamines General Statement 4:00.) The drug also should not be used for topical self-medication in the management of varicella (chickenpox) or measles without first consulting a clinician.
The nasal inhaler delivers about 50 mcg of fluticasone propionate per The usual adult oral dosage of diphenhydramine hydrochloride is 25-50 mg 3 metered spray. The commercially available preparation delivers about 120 or 4 times daily at 4- to 6-hour intervals, not to exceed 300 mg in 24 metered sprays per 16-g bottle; the container should be discarded after the hours. labeled number of actuations have been used since the correct dose per
inhalation cannot be assured if used for additional doses. Dosage of The usual adult IM or IV dose of diphenhydramine hydrochloride is 10-50 mg; intranasal fluticasone propionate should be adjusted according to in a few patients, up to 100 mg may be required. Some experts recommend a individual requirements and response; the lowest effective dosage should be dose of 25-50 mg.
The rate of IV administration should not exceed 25 used in order to minimize potential systemic effects of the drug. (See mg/minute. Cautions: Hypothalamic-Pituitary-Adrenal (HPA) Axis Suppression.) The
maximum daily dosage of intranasal fluticasone propionate should not exceed The maximum adult IM or IV dosage of diphenhydramine hydrochloride is 400 100 mcg in each nostril (total of 200 mcg daily). mg daily.
In patients with chronic renal impairment (creatinine clearance of 30 mL/minute or less), both oral bioavailability and peak plasma concentrations of loratadine and desloratadine may be increased compared with individuals with normal renal function. However, elimination half-lives of the drug and its active metabolite appear to be similar to those of individuals with normal renal function. Patients with renal impairment receiving loratadine for self-medication should be advised to consult a clinician before initiating therapy, since a different dosage may be recommended.
Therapy with loratadine conventional or orally disintegrating tablets or oral solution should be initiated at a dosage of 10 mg every other day in adults and children 6 years of age and older with a glomerular filtration rate less than 30 mL/minute and at a dosage of 5 mg every other day in children 2-5 years of age with renal insufficiency. In addition, therapy with the commercially available tablets containing loratadine in fixed combination with pseudoephedrine sulfate should be initiated in adults and children 12 years of age and older with a glomerular filtration rate less than 30 mL/minute at a dosage of 5 mg once daily when the 12-hour formulation is used or at a dosage of 10 mg every other day when the 24-hour formulation is used, since clearance of both loratadine and pseudoephedrine are decreased in such patients. Hemodialysis does not appear to affect the pharmacokinetics of loratadine or desloratadine.
The pharmacokinetics of loratadine and its active metabolite also may be altered in patients with hepatic impairment and dosage adjustment may be necessary. Therefore, patients with hepatic impairment receiving loratadine for self-medication should be advised to consult a clinician before initiating therapy, since a different dosage may be recommended. Therapy with loratadine conventional or orally disintegrating tablets or oral solution should be initiated at a dosage of 10 mg every other day in adults and children 6 years of age and older with hepatic failure and at a dosage of 5 mg every other day in children 2-5 years of age with hepatic failure.
Since fixed-ratio combination preparations do not permit individual titration of dosages, and clearance of loratadine is decreased more substantially than that of pseudoephedrine sulfate in patients with hepatic impairment, the manufacturer recommends that tablets containing loratadine in fixed combination with pseudoephedrine sulfate generally not be used in such patients.
Adjustment of fexofenadine hydrochloride dosage may be necessary in patients with renal impairment. Peak plasma fexofenadine concentrations increased by 87 or 111%, and elimination half-life increased by 59 or 72% in patients with mild (e.g., creatinine clearance of 41-80 mL/minute) or severe (creatinine clearance of 11-40 mL/minute) renal impairment, respectively, when compared with those observed in healthy individuals. In addition, peak plasma fexofenadine concentration increased by 82% and elimination half-life increased by 31% in those on hemodialysis (creatinine clearance of 10 mL/minute or less) compared with healthy individuals.
The manufacturer states that adults and children 12 years of age and older with impaired renal function or those on hemodialysis should receive an initial fexofenadine hydrochloride dosage of 60 mg daily (either given alone or in fixed combination with 120 mg of pseudoephedrine hydrochloride (Allegra-D(R) 12 Hour)). The fixed-combination preparation containing 180 mg of fexofenadine hydrochloride and 240 mg of pseudoephedrine hydrochloride (Allegra-D(R) 24 Hour) generally should be avoided in patients with renal impairment because of a possible risk of accumulation of pseudoephedrine.
Children 6 to younger than 12 years of age with impaired renal function should receive an initial fexofenadine hydrochloride dosage of 30 mg daily.
Since the pharmacokinetics of fexofenadine do not appear to be altered in patients with hepatic impairment, the manufacturer states that dosage adjustment is not necessary in such patients. The manufacturer of Allegra-D(R) 12 Hour and Allegra-D(R) 24 Hour does not make specific recommendations for dosage adjustment in patients with hepatic impairment, although it is not known if pharmacokinetics of pseudoephedrine are altered in patients with hepatic impairment.
The manufacturer states that patients 12 years of age or older who have impaired renal function (e.g., creatinine clearance of 11-31 mL/minute) or hepatic impairment or who are undergoing hemodialysis (creatinine clearance of less than 7 mL/minute), should receive a cetirizine hydrochloride dosage of 5 mg daily. The manufacturer also states that children 6-11 years of age with impaired renal or hepatic function should use the lower recommended dosage (5 mg once daily). The manufacturer states that use of cetirizine hydrochloride in children younger than 6 years of age with impaired renal or hepatic function is not recommended because administration of doses smaller than 2.5
mg is difficult and not reliable, and pharmacokinetic data are lacking in this patient population.
When extended-release tablets of cetirizine hydrochloride in fixed combination with pseudoephedrine hydrochloride are used in patients 12 years of age or older who have impaired renal function (i.e., creatinine clearance of 11-31 mL/minute) or hepatic impairment or who are undergoing hemodialysis (creatinine clearance of less than 7 mL/minute), the recommended cetirizine hydrochloride dosage is 5 mg once daily.
When diphenhydramine was available only by prescription, the prescribing information for the drug indicated a usual oral diphenhydramine hydrochloride dosage for children weighing more than 9.1 kg of 12.5-25 mg 3 or 4 times daily at 4- to 6-hour intervals and for children weighing 9.1
kg or less an oral diphenhydramine hydrochloride dosage of 6.25-12.5 mg 3 or 4 times daily at 4- to 6-hour intervals.
However, these dosage recommendations are not included in the current labeling of nonprescription oral diphenhydramine preparations, and clinicians should use caution when considering use of nonprescription oral diphenhydramine in children younger than 4 years of age. (See Cautions: Pediatric Precautions.)
Alternatively, for oral, deep IM, or IV therapy, children (other than premature or full-term neonates) may be given 5 mg/kg daily or 150 mg/m2 daily divided in 4 doses; some experts recommend a dosage of 1-2 mg/kg daily. The rate of IV administration should not exceed 25 mg/minute.
The maximum oral, IM, or IV dosage of diphenhydramine hydrochloride in children older than 1 month of age is 300 mg daily.
For temporary relief of pruritus and pain associated with various skin conditions in adults and children 2 years of age or older, creams, lotions, or solutions containing 1-2% diphenhydramine hydrochloride are applied to the affected areas 3 or 4 times daily or as directed by a clinician; topical diphenhydramine should not be used more often than directed.
If the condition worsens, or if symptoms persist for longer than 7 days or resolve and then recur within a few days, topical therapy with diphenhydramine hydrochloride should be discontinued and a clinician consulted; the possibility of sensitization by, or hypersensitivity to, the drug should be considered.
Topical preparations containing diphenhydramine hydrochloride should not be used on large areas of the body or concomitantly with other preparations containing the antihistamine, including those used orally, since increased serum concentrations of diphenhydramine may occur that can result in systemic toxicity. (See Acute Toxicity: Manifestations, in the Antihistamines General Statement 4:00.) The drug also should not be used for topical self-medication in the management of varicella (chickenpox) or measles without first consulting a clinician.
Diphenhydramine hydrochloride usually is administered orally. Diphenhydramine citrate usually is administered orally. When oral therapy is not feasible, diphenhydramine hydrochloride may be given by deep IM or, preferably, IV injection.
The drug should not be given subcutaneously, intradermally, or perivascularly because of its irritating effects; local necrosis has been reported following subcutaneous or intradermal administration of parenteral diphenhydramine. IV use of the drug in a home-care setting should be employed under careful supervision. Use of diphenhydramine for local anesthesia via local infiltration is discouraged because of the risk of local tissue necrosis.
Diphenhydramine hydrochloride should not be given to premature or full-term neonates. (See Cautions: Pediatric Precautions.) For the temporary relief of pruritus associated with various skin conditions and disorders, diphenhydramine hydrochloride-containing preparations are applied topically in the form of a cream, lotion, or topical solution. The possibility of clinically important percutaneous absorption of the drug following topical application should be considered.
(See Cautions.) Fluticasone propionate is administered by nasal inhalation using a Loratadine is administered orally. Loratadine conventional tablets, orally disintegrating tablets, and the commercially available tablets containing metered-dose nasal spray pump. Patients should be instructed carefully in the drug in fixed combination with pseudoephedrine sulfate can be the use of the nasal spray pump, including the need to prime the pump prior administered without regard to meals.
to first use or after a period of nonuse (i.e., 1 week or more). To obtain optimum results, patients also should be given a copy of the patient Although the oral bioavailability of loratadine is increased when the drug instructions provided by the manufacturer. is administered as the orally disintegrating tablet without water, the bioavailability of the active metabolite desloratadine Prior to administration of fluticasone propionate nasal spray, patients (descarboethoxyloratadine) is unaffected, and the manufacturers state that should clear their nasal passages; administration of a topical nasal decongestant about 5-15 minutes before intranasal corticosteroid the orally disintegrating tablets can be administered with or without administration may be useful during the first 2 or 3 days of therapy in water.
The orally disintegrating tablets are administered by placing a patients with blocked nasal passages to ensure adequate penetration of the tablet on the tongue, where it disintegrates within a few seconds, and then drug. Prior to initial use, the nasal inhaler must be primed. Patients subsequently swallowing with or without water.
Tablets containing loratadine in fixed combination with pseudoephedrine sulfate should be should tilt the head slightly forward, insert the nasal adapter into one nostril, and point the tip of the adapter toward the inflamed nasal swallowed intact and patients should be instructed not to break, chew, or dissolve such tablets. Patients also should be instructed to take turbinates and away from the nasal septum. For maximum therapeutic effect and to ensure adequate penetration of the drug, patients should pump the Claritin-D(R) 24 Hour extended-release tablets with a full glass of water.
drug into one nostril while holding the other nostril closed and should concurrently inspire through the nose. This procedure is then repeated for the other nostril. If sneezing occurs during drug administration, patients should wait until sneezing has stopped, then clear the nasal passages and repeat administration of the dose.
The manufacturer recommends cleaning of the nasal spray adapter and/or pump at least once weekly. After removing the nasal adapter and dust cap, these pieces should be rinsed in warm water and dried thoroughly. If the nasal adapter becomes clogged, the piece should be soaked in warm water; cleaning by inserting a sharp object into the piece is not recommended.
Fexofenadine hydrochloride is administered orally. The manufacturer states that when fexofenadine hydrochloride is given alone (i.e., not in fixed combination with pseudoephedrine hydrochloride) the drug may be given without regard to meals. Since absorption and peak plasma concentrations of fexofenadine are decreased by concomitant administration of an aluminum and magnesium hydroxides antacid (Maalox(R)) (see Pharmacokinetics: Absorption and see Drug Interactions: Antacids), the manufacturer recommends that the drug not be taken closely in time with an antacid containing aluminum and magnesium.
Since food appears to substantially affect the rate and extent of absorption of fexofenadine hydrochloride when administered as the extended-release tablets of the drug in fixed combination with pseudoephedrine hydrochloride, the manufacturer states that such extended-release tablets should be administered on an empty stomach with water. (See Pharmacokinetics: Absorption and see Drug Interactions: Fruit Juices.) Extended-release tablets containing fexofenadine hydrochloride in fixed combination with pseudoephedrine hydrochloride should be swallowed intact, and patients should be instructed not to break, crush, or chew such tablets. Cetirizine is administered orally.
Cetirizine oral solution (syrup) should be administered using the measuring device (i.e., cup) provided by the manufacturer. Cetirizine chewable tablets may be administered with or without water. Tablets containing cetirizine hydrochloride in fixed combination with pseudoephedrine hydrochloride should be swallowed intact and patients should be instructed not to break or chew such tablets.
The manufacturer states that the time of administration of cetirizine may be adjusted for individual patient requirements. Although food may decrease peak plasma concentrations of cetirizine and lengthen the time to achievement of peak plasma concentrations, the manufacturer states that cetirizine may be administered without regard to food because food does not affect the extent of absorption of the drug when administered as conventional or chewable tablets. The oral bioavailability of cetirizine hydrochloride conventional tablets is comparable to that of the oral solution and to that of the chewable tablets (administered with or without water).
The drug should not be given subcutaneously, intradermally, or perivascularly because of its irritating effects; local necrosis has been reported following subcutaneous or intradermal administration of parenteral diphenhydramine. IV use of the drug in a home-care setting should be employed under careful supervision. Use of diphenhydramine for local anesthesia via local infiltration is discouraged because of the risk of local tissue necrosis.
Diphenhydramine hydrochloride should not be given to premature or full-term neonates. (See Cautions: Pediatric Precautions.) For the temporary relief of pruritus associated with various skin conditions and disorders, diphenhydramine hydrochloride-containing preparations are applied topically in the form of a cream, lotion, or topical solution. The possibility of clinically important percutaneous absorption of the drug following topical application should be considered.
(See Cautions.) Fluticasone propionate is administered by nasal inhalation using a Loratadine is administered orally. Loratadine conventional tablets, orally disintegrating tablets, and the commercially available tablets containing metered-dose nasal spray pump. Patients should be instructed carefully in the drug in fixed combination with pseudoephedrine sulfate can be the use of the nasal spray pump, including the need to prime the pump prior administered without regard to meals.
to first use or after a period of nonuse (i.e., 1 week or more). To obtain optimum results, patients also should be given a copy of the patient Although the oral bioavailability of loratadine is increased when the drug instructions provided by the manufacturer. is administered as the orally disintegrating tablet without water, the bioavailability of the active metabolite desloratadine Prior to administration of fluticasone propionate nasal spray, patients (descarboethoxyloratadine) is unaffected, and the manufacturers state that should clear their nasal passages; administration of a topical nasal decongestant about 5-15 minutes before intranasal corticosteroid the orally disintegrating tablets can be administered with or without administration may be useful during the first 2 or 3 days of therapy in water.
The orally disintegrating tablets are administered by placing a patients with blocked nasal passages to ensure adequate penetration of the tablet on the tongue, where it disintegrates within a few seconds, and then drug. Prior to initial use, the nasal inhaler must be primed. Patients subsequently swallowing with or without water.
Tablets containing loratadine in fixed combination with pseudoephedrine sulfate should be should tilt the head slightly forward, insert the nasal adapter into one nostril, and point the tip of the adapter toward the inflamed nasal swallowed intact and patients should be instructed not to break, chew, or dissolve such tablets. Patients also should be instructed to take turbinates and away from the nasal septum. For maximum therapeutic effect and to ensure adequate penetration of the drug, patients should pump the Claritin-D(R) 24 Hour extended-release tablets with a full glass of water.
drug into one nostril while holding the other nostril closed and should concurrently inspire through the nose. This procedure is then repeated for the other nostril. If sneezing occurs during drug administration, patients should wait until sneezing has stopped, then clear the nasal passages and repeat administration of the dose.
The manufacturer recommends cleaning of the nasal spray adapter and/or pump at least once weekly. After removing the nasal adapter and dust cap, these pieces should be rinsed in warm water and dried thoroughly. If the nasal adapter becomes clogged, the piece should be soaked in warm water; cleaning by inserting a sharp object into the piece is not recommended.
Fexofenadine hydrochloride is administered orally. The manufacturer states that when fexofenadine hydrochloride is given alone (i.e., not in fixed combination with pseudoephedrine hydrochloride) the drug may be given without regard to meals. Since absorption and peak plasma concentrations of fexofenadine are decreased by concomitant administration of an aluminum and magnesium hydroxides antacid (Maalox(R)) (see Pharmacokinetics: Absorption and see Drug Interactions: Antacids), the manufacturer recommends that the drug not be taken closely in time with an antacid containing aluminum and magnesium.
Since food appears to substantially affect the rate and extent of absorption of fexofenadine hydrochloride when administered as the extended-release tablets of the drug in fixed combination with pseudoephedrine hydrochloride, the manufacturer states that such extended-release tablets should be administered on an empty stomach with water. (See Pharmacokinetics: Absorption and see Drug Interactions: Fruit Juices.) Extended-release tablets containing fexofenadine hydrochloride in fixed combination with pseudoephedrine hydrochloride should be swallowed intact, and patients should be instructed not to break, crush, or chew such tablets. Cetirizine is administered orally.
Cetirizine oral solution (syrup) should be administered using the measuring device (i.e., cup) provided by the manufacturer. Cetirizine chewable tablets may be administered with or without water. Tablets containing cetirizine hydrochloride in fixed combination with pseudoephedrine hydrochloride should be swallowed intact and patients should be instructed not to break or chew such tablets.
The manufacturer states that the time of administration of cetirizine may be adjusted for individual patient requirements. Although food may decrease peak plasma concentrations of cetirizine and lengthen the time to achievement of peak plasma concentrations, the manufacturer states that cetirizine may be administered without regard to food because food does not affect the extent of absorption of the drug when administered as conventional or chewable tablets. The oral bioavailability of cetirizine hydrochloride conventional tablets is comparable to that of the oral solution and to that of the chewable tablets (administered with or without water).
| DRUG LABEL | DOSING TYPE | DOSING INSTRUCTIONS |
|---|---|---|
| EQ ALLERGY RELIEF 1 MG/ML SOLN | Maintenance | Adults take 10 milliliters (10 mg) by oral route once daily |
| EQ ALLERGY RELIEF 25 MG CAP | Maintenance | Adults take 1 capsule (25 mg) by oral route every 4-6 hours as needed |
| EQ ALLERGY RELIEF 50 MCG SPRAY | Maintenance | Adults spray 1 - 2 sprays (50 - 100 mcg) in each nostril by intranasal route once daily as needed |
| EQ ALLERGY RELIEF 180 MG TAB | Maintenance | Adults take 1 tablet (180 mg) by oral route once daily |
| EQ ALLERGY RELIEF 10 MG TABLET | Maintenance | Adults take 1 tablet (10 mg) by oral route once daily |
| EQ ALLERGY (LORAT) 10 MG TAB | Maintenance | Adults take 1 tablet (10 mg) by oral route once daily |
| EQ ALLERGY RELIEF 25 MG TABLET | Maintenance | Adults take 1 tablet (25 mg) by oral route every 4-6 hours as needed |
| DRUG LABEL | DOSING TYPE | DOSING INSTRUCTIONS |
|---|---|---|
| LORATADINE 10 MG TABLET | Maintenance | Adults take 1 tablet (10 mg) by oral route once daily |
| CETIRIZINE HCL 1 MG/ML SOLN | Maintenance | Adults take 10 milliliters (10 mg) by oral route once daily |
| FEXOFENADINE HCL 180 MG TABLET | Maintenance | Adults take 1 tablet (180 mg) by oral route once daily |
| CETIRIZINE HCL 10 MG TABLET | Maintenance | Adults take 1 tablet (10 mg) by oral route once daily |
| FLUTICASONE PROP 50 MCG SPRAY | Maintenance | Adults spray 1 - 2 sprays (50 - 100 mcg) in each nostril by intranasal routeonce daily as needed |
| DIPHENHYDRAMINE 25 MG TABLET | Maintenance | Adults take 1 tablet (25 mg) by oral route every 4-6 hours as needed |
| CHILD CETIRIZINE HCL 1 MG/ML | Maintenance | Adults take 10 milliliters (10 mg) by oral route once daily |
| DIPHENHYDRAMINE 25 MG CAPSULE | Maintenance | Adults take 1 capsule (25 mg) by oral route every 4-6 hours as needed |
| DIPHENHYDRAMINE 25 MG CAPLET | Maintenance | Adults take 1 tablet (25 mg) by oral route every 4-6 hours as needed |
| CETIRIZINE HCL 1 MG/ML SYRUP | Maintenance | Adults take 10 milliliters (10 mg) by oral route once daily |
| GNP LORATADINE 10 MG TABLET | Maintenance | Adults take 1 tablet (10 mg) by oral route once daily |
| EQL FLUTICASONE PROP 50 MCG SP | Maintenance | Adults spray 1 - 2 sprays (50 - 100 mcg) in each nostril by intranasal routeonce daily as needed |
| GNP FLUTICASONE PROP 50 MCG SP | Maintenance | Adults spray 1 - 2 sprays (50 - 100 mcg) in each nostril by intranasal routeonce daily as needed |
| GNP FEXOFENADINE HCL 180 MG TB | Maintenance | Adults take 1 tablet (180 mg) by oral route once daily |
| EQ CHILD CETIRIZINE 1 MG/ML | Maintenance | Adults take 10 milliliters (10 mg) by oral route once daily |
| CVS FLUTICASONE PROP 50 MCG SP | Maintenance | Adults spray 1 - 2 sprays (50 - 100 mcg) in each nostril by intranasal routeonce daily as needed |
The following drug interaction information is available for ALLERGY RELIEF (cetirizine hcl):
There are 1 contraindications.
These drug combinations generally should not be dispensed or administered to the same patient. A manufacturer label warning that indicates the contraindication warrants inclusion of a drug combination in this category, regardless of clinical evidence or lack of clinical evidence to support the contraindication.
| Drug Interaction | Drug Names |
|---|---|
| Selected Antihistamines/Selected MAOIs SEVERITY LEVEL: 1-Contraindicated Drug Combination: This drug combination is contraindicated and generally should not be dispensed or administered to the same patient. MECHANISM OF ACTION: MAOIs prolong and intensify the effects of antihistamines.(1-6) CLINICAL EFFECTS: Concurrent use of antihistamines and a MAOI may result in severe hypotension.(1-6) PREDISPOSING FACTORS: None determined. PATIENT MANAGEMENT: Concurrent use of antihistamines and a MAOI is contraindicated.(1-6) DISCUSSION: MAOIs may prolong and intensify the effects of antihistamines, resulting in severe hypotension.(1-6) A case report describes a patient having cyproheptadine added to their phenelzine therapy in an attempt to relieve the patients anorgasmia. The patient began to suddenly experience visual hallucination after taking the cyproheptadine for two months. Once the medication was terminated, the hallucinations stopped occurring within 48 hours.(7) Methylene blue, when administered intravenously, has been shown to reach sufficient concentrations to be a potent inhibitor of MAO-A.(8,9) |
FURAZOLIDONE, MARPLAN, MATULANE, METHYLENE BLUE, NARDIL, PARNATE, PHENELZINE SULFATE, PROCARBAZINE HCL, PROVAYBLUE, TRANYLCYPROMINE SULFATE |
There are 9 severe interactions.
These drug interactions can produce serious consequences in most patients. Actions required for severe interactions include, but are not limited to, discontinuing one or both agents, adjusting dosage, altering administration scheduling, and providing additional patient monitoring. Review the full interaction monograph for more information.
| Drug Interaction | Drug Names |
|---|---|
| Selected Steroids/Antiretroviral CYP3A4 Inhibitors SEVERITY LEVEL: 2-Severe Interaction: Action is required to reduce the risk of severe adverse interaction. MECHANISM OF ACTION: Antiretroviral CYP3A4 inhibitors may inhibit the metabolism of corticosteroids metabolized by CYP3A4. Dexamethasone may induce metabolism of agents that are substrates of CYP3A4.(1-13,50) CLINICAL EFFECTS: Concurrent use of antiretroviral CYP3A4 inhibitors may result in increased systemic exposure to and effects from corticosteroids metabolized by CYP3A4, including Cushing's syndrome and adrenal suppression. Concurrent dexamethasone may result in decreased levels and effectiveness of CYP3A4 substrates. PREDISPOSING FACTORS: None determined. PATIENT MANAGEMENT: If possible, avoid concurrent therapy of corticosteroids metabolized by CYP3A4 with antiretroviral CYP3A4 inhibitors. Alternative corticosteroids that are less affected by CYP3A4 inhibitors should be considered, like beclomethasone, prednisone, and prednisolone. If concurrent therapy is warranted, patients should be closely monitored for systemic effects. The corticosteroid may need to be discontinued. Patients receiving concurrent therapy with dexamethasone and substrates of CYP3A4 should also be monitored for decreased effectiveness of the CYP3A4 substrate. The manufacturers of nasal fluticasone(14-16) and fluticasone for inhalation(17) state that concurrent use of fluticasone and atazanavir, indinavir, nelfinavir, ritonavir or saquinavir is not recommended. The US manufacturers of atazanavir,(1) fosamprenavir,(5) indinavir(6) and nelfinavir(8) recommend caution with concurrent use of inhaled or nasal fluticasone. Consider alternatives to fluticasone if long-term use is required. DISCUSSION: In a study, boceprevir (800 mg TID for 7 days) increased the area-under-curve (AUC) of a single dose of prednisone (40 mg) by 22%. The maximum concentration (Cmax) and AUC of prednisolone increased by 16% and 37%, respectively.(2) A study of 14 healthy adults found that concurrent use of ketoconazole with ciclesonide increased the AUC of ciclesonide's active metabolite, des-ciclesonide, by approximately 3.6-fold at steady state, while levels of ciclesonide remained unchanged. However, the study concluded that no dosage adjustments were required because ciclesonide has a very low potential to cause side effects.(18) A study in 18 healthy subjects examined the effects of ritonavir (100 mg twice daily) on fluticasone nasal spray (200 mcg daily). In most subjects, fluticasone was undetectable (<10 pg/ml) when administered alone. In subjects in whom fluticasone was detectable when given alone, Cmax and area-under-curve AUC averaged 11.9 pg/ml and 8.43 pg x hr/ml, respectively. With concurrent ritonavir, fluticasone Cmax and AUC increased to 318 pg/ml and 3102.6 pg x hr/ml, respectively.(7,11,14) This reflects increases in Cmax and AUC by 25-fold and 350-fold, respectively.(3) The cortisol AUC decreased by 86%.(6,14-16) In a study in 10 healthy subjects, ritonavir (200 mg twice daily for 4 and 14 days) increased the AUC of a single dose of prednisolone by 1.41-fold and 1.30-fold, respectively, after 4 days and 14 days of ritonavir.(19) There have been several case reports of Cushing's syndrome in patients treated concurrently with ritonavir and inhaled budesonide,(19-20) dexamethasone,(22) injectable triamcinolone,(23-26) nasal fluticasone.(28-46) Hepatitis has also been reported with concurrent budesonide and ritonavir.(47) In a study in 9 healthy subjects, mibefradil (50 mg once daily for 3 days) increased the AUC, Cmax, and elimination half-life of methylprednisolone by 3.8-fold, 1.8-fold, and 2.7-fold, respectively.(48) In a study in 8 healthy subjects, following nefazodone administration the following changes were seen with methylprednisolone: mean (+/-SD) area under the concentration-time curve was significantly higher (1393 +/- 343 vs. 2966 +/- 928 ug*h/L; P < 0.005), apparent clearance was lower (28.7 +/- 7.2 vs. 14.6 +/- 7.8 L/h; P < 0.02) and the terminal elimination half-life was longer (2.28 +/- 0.49 vs. 3.32 +/- 0.95 hours; P < 0.02).(49) Selected steroids metabolized by CYP3A4 are linked to this monograph.(50) Selected CYP3A4 inhibitors and substrates linked to this monograph include: atazanavir, cobicistat, darunavir, fosamprenavir, indinavir, lenacapavir, lopinavir, nelfinavir, saquinavir, and tipranavir.(50) |
APTIVUS, ATAZANAVIR SULFATE, DARUNAVIR, EVOTAZ, FOSAMPRENAVIR CALCIUM, GENVOYA, KALETRA, LOPINAVIR-RITONAVIR, PREZCOBIX, PREZISTA, REYATAZ, STRIBILD, SUNLENCA, SYMTUZA, VIRACEPT, YEZTUGO |
| Solid Oral Potassium Tablets/Anticholinergics SEVERITY LEVEL: 2-Severe Interaction: Action is required to reduce the risk of severe adverse interaction. MECHANISM OF ACTION: Concentrated potassium may damage the lining of the GI tract. Anticholinergics delay gastric emptying, resulting in the potassium product remaining in the gastrointestinal tract for a longer period of time.(1-16) CLINICAL EFFECTS: Use of solid oral dosage forms of potassium in patients treated with anticholinergics may result in gastrointestinal erosions, ulcers, stenosis and bleeding.(1-16) PREDISPOSING FACTORS: Diseases or conditions which may increase risk for GI damage include: preexisting dysphagia, strictures, cardiomegaly, diabetic gastroparesis, elderly status, or insufficient oral intake to allow dilution of potassium.(1-10,21) Other drugs which may add to risk for GI damage include: nonsteroidal anti-inflammatory drugs (NSAIDs), bisphosphonates, or tetracyclines.(21) PATIENT MANAGEMENT: Regulatory agency and manufacturer recommendations regarding this interaction: - In the US, all solid oral dosage forms (including tablets and extended release capsules) of potassium are contraindicated in patients receiving anticholinergics at sufficient dosages to result in systemic effects.(2-8) Patients receiving such anticholinergic therapy should use a liquid form of potassium chloride.(2) - In Canada, solid oral potassium is contraindicated in any patient with a cause for arrest or delay in tablet/capsule passage through the gastrointestinal tract and the manufacturers recommend caution with concurrent anticholinergic medications.(1,9-10) Evaluate each patient for predisposing factors which may increase risk for GI damage. In patients with multiple risk factors for harm, consider use of liquid potassium supplements, if tolerated. For patients receiving concomitant therapy, assure any potassium dose form is taken after meals with a large glass of water or other fluid. To decrease potassium concentration in the GI tract, limit each dose to 20 meq; if more than 20 meq daily is required, give in divided doses.(2) If concurrent therapy is warranted, monitor patients receiving concurrent therapy for signs of blood loss, including decreased hemoglobin, hematocrit, fecal occult blood, and/or decreased blood pressure and promptly evaluate patients with any symptoms. Patients should be instructed to immediately report any difficulty swallowing, abdominal pain, distention, severe vomiting, or gastrointestinal bleeding. Instruct patients to report any signs and symptoms of bleeding, such as unusual bleeding from the gums or nose; unusual bruising; red or black, tarry stools; red, pink or dark brown urine; acute abdominal or joint pain and/or swelling. DISCUSSION: In clinical trials, there was a higher incidence of gastric and duodenal lesions in patients receiving a high dose of a wax-matrix controlled-release formulation with a concurrent anticholinergic agent. Some lesions were asymptomatic and not accompanied by bleeding, as shown by a lack of positive Hemoccult tests.(1-17) Several studies suggest that the incidence of gastric and duodenal lesions may be less with the microencapsulated formulation of potassium chloride.(14-17) |
KLOR-CON 10, KLOR-CON 8, KLOR-CON M10, KLOR-CON M15, KLOR-CON M20, POTASSIUM CHLORIDE, POTASSIUM CITRATE ER, UROCIT-K |
| Solid Oral Potassium Capsules/Anticholinergics SEVERITY LEVEL: 2-Severe Interaction: Action is required to reduce the risk of severe adverse interaction. MECHANISM OF ACTION: Concentrated potassium may damage the lining of the GI tract. Anticholinergics delay gastric emptying, resulting in the potassium product remaining in the gastrointestinal tract for a longer period of time.(1-16)) CLINICAL EFFECTS: Use of solid oral dosage forms of potassium in patients treated with anticholinergics may result in gastrointestinal erosions, ulcers, stenosis and bleeding.(1-16) PREDISPOSING FACTORS: Diseases or conditions which may increase risk for GI damage include: preexisting dysphagia, strictures, cardiomegaly, diabetic gastroparesis, elderly status, or insufficient oral intake to allow dilution of potassium.(1-10,21) Other drugs which may add to risk for GI damage include: nonsteroidal anti-inflammatory drugs (NSAIDs), bisphosphonates, or tetracyclines.(21) PATIENT MANAGEMENT: Regulatory agency and manufacturer recommendations regarding this interaction: - In the US, all solid oral dosage forms (including tablets and extended release capsules) of potassium are contraindicated in patients receiving anticholinergics at sufficient dosages to result in systemic effects.(2-8) Patients receiving such anticholinergic therapy should use a liquid form of potassium chloride.(2) - In Canada, solid oral potassium is contraindicated in any patient with a cause for arrest or delay in tablet/capsule passage through the gastrointestinal tract and the manufacturers recommend caution with concurrent anticholinergic medications.(1,9-10) Evaluate each patient for predisposing factors which may increase risk for GI damage. In patients with multiple risk factors for harm, consider use of liquid potassium supplements, if tolerated. For patients receiving concomitant therapy, assure any potassium dose form is taken after meals with a large glass of water or other fluid. To decrease potassium concentration in the GI tract, limit each dose to 20 meq; if more than 20 meq daily is required, give in divided doses.(2) If concurrent therapy is warranted, monitor patients receiving concurrent therapy for signs of blood loss, including decreased hemoglobin, hematocrit, fecal occult blood, and/or decreased blood pressure and promptly evaluate patients with any symptoms. Patients should be instructed to immediately report any difficulty swallowing, abdominal pain, distention, severe vomiting, or gastrointestinal bleeding. Instruct patients to report any signs and symptoms of bleeding, such as unusual bleeding from the gums or nose; unusual bruising; red or black, tarry stools; red, pink or dark brown urine; acute abdominal or joint pain and/or swelling. DISCUSSION: In clinical trials, there was a higher incidence of gastric and duodenal lesions in patients receiving a high dose of a wax-matrix controlled-release formulation with a concurrent anticholinergic agent. The lesions were asymptomatic and not accompanied by bleeding, as shown by a lack of positive Hemoccult tests.(1-17) Several studies suggest that the incidence of gastric and duodenal lesions may be less with the microencapsulated formulation of potassium chloride.(14-17) |
POTASSIUM CHLORIDE |
| Radioactive Iodide/Agents that Affect Iodide SEVERITY LEVEL: 2-Severe Interaction: Action is required to reduce the risk of severe adverse interaction. MECHANISM OF ACTION: Many compounds can affect iodide protein binding and alter iodide pharmacokinetics and pharmacodynamics.(1) CLINICAL EFFECTS: Compounds that affect iodide pharmacokinetics and pharmacodynamics may impact the effectiveness of radioactive iodide.(1) PREDISPOSING FACTORS: Compounds that affect iodide pharmacokinetics and pharmacodynamics are expected to have the most impact during therapy using radioactive iodide. Diagnostic procedures would be expected to be impacted less. PATIENT MANAGEMENT: Discuss the use of agents that affect iodide pharmacokinetics and pharmacodynamics with the patient's oncologist.(1) Because indocyanine green contains sodium iodide, the iodine-binding capacity of thyroid tissue may be reduced for at least one week following administration. Do not perform radioactive iodine uptake studies for at least one week following administration of indocyanine green.(2) The manufacturer of iopamidol states administration may interfere with thyroid uptake of radioactive iodine and decrease therapeutic and diagnostic efficacy. Avoid thyroid therapy or testing for up to 6 weeks post administration of iopamidol.(3) DISCUSSION: Many agents interact with radioactive iodine. The average duration of effect is: anticoagulants - 1 week antihistamines - 1 week anti-thyroid drugs, e.g: carbimazole, methimazole, propylthiouracil - 3-5 days corticosteroids - 1 week iodide-containing medications, e.g: amiodarone - 1-6 months expectorants - 2 weeks Lugol solution - 3 weeks saturated solution of potassium iodine - 3 weeks vitamins - 10-14 days iodide-containing X-ray contrast agents - up to 1 year lithium - 4 weeks phenylbutazone - 1-2 weeks sulfonamides - 1 week thyroid hormones (natural or synthetic), e.g.: thyroxine - 4 weeks tri-iodothyronine - 2 weeks tolbutamide - 1 week topical iodide - 1-9 months (1) |
ADREVIEW, JEANATOPE, MEGATOPE, SODIUM IODIDE I-123, VOLUMEX |
| Clozapine/Anticholinergics SEVERITY LEVEL: 2-Severe Interaction: Action is required to reduce the risk of severe adverse interaction. MECHANISM OF ACTION: Clozapine has potent anticholinergic properties and inhibits serotonin receptors, including 5-HT3.(1-4) Both of these properties may cause inhibition of gastrointestinal (GI) smooth muscle contraction, resulting in decreased peristalsis.(3,4) These effects may be compounded by concurrent use of anticholinergic agents.(1-6) CLINICAL EFFECTS: Concurrent use of clozapine with other anticholinergic agents may increase the risk of constipation (common) and serious bowel complications (uncommon), including complete bowel obstruction, fecal impaction, paralytic ileus and intestinal ischemia or infarction.(1-6) PREDISPOSING FACTORS: The risk for serious bowel complications is higher with increasing age, higher frequency of constipation, and in patients on higher doses of clozapine or multiple anticholinergic agents.(1,5) PATIENT MANAGEMENT: Avoid the use of other anticholinergic agents with clozapine.(1-6) If concurrent use is necessary, evaluate the patient's bowel function regularly. Monitor for symptoms of constipation and GI hypomotility, including having bowel movements less than three times weekly or less than usual, difficulty having a bowel movement or passing gas, nausea, vomiting, and abdominal pain or distention.(2) Consider a prophylactic laxative in those with a history of constipation or bowel obstruction.(2) Review patient medication list for other anticholinergic agents. When possible, decrease the dosage or number of prescribed anticholinergic agents, particularly in the elderly. Counsel the patient about the importance of maintaining adequate hydration. Encourage regular exercise and eating a high-fiber diet.(2) DISCUSSION: In a prospective cohort study of 26,720 schizophrenic patients in the Danish Central Psychiatric Research Registry, the odds ratio (OR) for ileus was 1.99 with clozapine and 1.48 with anticholinergics. The OR for fatal ileus was 6.73 with clozapine and 5.88 with anticholinergics. Use of anticholinergics with 1st generation antipsychotics (FGA) increased the risk of ileus compare to FGA alone, but this analysis was not done with clozapine.(5) A retrospective cohort study of 24,970 schizophrenic patients from the Taiwanese National Health Insurance Research Database found that the hazard ratio (HR) for clozapine-induced constipation increased from 1.64 when clozapine is used alone, to 2.15 when used concomitantly with anticholinergics. However, there was no significant difference in the HR for ileus when clozapine is used with and without anticholinergics (1.95 and 2.02, respectively).(6) In the French Pharmacovigilance Database, 7 of 38 cases of antipsychotic-associated ischemic colitis or intestinal necrosis involved clozapine, and 5 of these cases involved use of concomitant anticholinergic agents. Three patients died, one of whom was on concomitant anticholinergics.(3) In a case series, 4 of 9 cases of fatal clozapine-associated GI dysfunction involved concurrent anticholinergic agents.(4) |
CLOZAPINE, CLOZAPINE ODT, CLOZARIL, VERSACLOZ |
| Eluxadoline/Anticholinergics; Opioids SEVERITY LEVEL: 2-Severe Interaction: Action is required to reduce the risk of severe adverse interaction. MECHANISM OF ACTION: Eluxadoline is a mixed mu-opioid and kappa-opioid agonist and delta-opioid antagonist and may alter or slow down gastrointestinal transit.(1) CLINICAL EFFECTS: Constipation related adverse events that sometimes required hospitalization have been reported, including the development of intestinal obstruction, intestinal perforation, and fecal impaction.(1) PREDISPOSING FACTORS: None determined. PATIENT MANAGEMENT: Avoid use with other drugs that may cause constipation. If concurrent use is necessary, evaluate the patient's bowel function regularly. Monitor for symptoms of constipation and GI hypomotility, including having bowel movements less than three times weekly or less than usual, difficulty having a bowel movement or passing gas, nausea, vomiting, and abdominal pain or distention.(1) Instruct patients to stop eluxadoline and immediately contact their healthcare provider if they experience severe constipation. Loperamide may be used occasionally for acute management of severe diarrhea, but must be discontinued if constipation develops.(1) DISCUSSION: In phase 3 clinical trials, constipation was the most commonly reported adverse reaction (8%). Approximately 50% of constipation events occurred within the first 2 weeks of treatment while the majority occurred within the first 3 months of therapy. Rates of severe constipation were less than 1% in patients receiving eluxadoline doses of 75 mg and 100 mg.(1) |
VIBERZI |
| Selected Steroids/Selected Strong CYP3A4 Inhibitors SEVERITY LEVEL: 2-Severe Interaction: Action is required to reduce the risk of severe adverse interaction. MECHANISM OF ACTION: Strong CYP3A4 inhibitors may inhibit the metabolism of corticosteroids metabolized by CYP3A4. CLINICAL EFFECTS: Concurrent use of strong CYP3A4 inhibitors may result in increased systemic exposure to and effects from corticosteroids metabolized by CYP3A4, including Cushing's syndrome and adrenal suppression. PREDISPOSING FACTORS: None determined. PATIENT MANAGEMENT: If possible, avoid concurrent therapy between corticosteroids metabolized by CYP3A4 and strong CYP3A4 inhibitors. Alternative corticosteroids that are less affected by CYP3A4 inhibitors should be considered, like beclomethasone, prednisone, and prednisolone. If concurrent therapy is warranted, patients should be closely monitored for systemic effects. The corticosteroid may need to be discontinued. DISCUSSION: In a study, boceprevir (800 mg TID for 7 days) increased the area-under-curve (AUC) of a single dose of prednisone (40 mg) by 22%. The maximum concentration (Cmax) and AUC of prednisolone increased by 16% and 37%, respectively.(3) A study of 14 healthy adults found that concurrent use of ketoconazole with ciclesonide increased the AUC of ciclesonide's active metabolite, des-ciclesonide, by approximately 3.6-fold at steady state, while levels of ciclesonide remained unchanged. However, the study concluded that no dosage adjustments were required because ciclesonide has a very low potential to cause side effects.(4) A study in 18 healthy subjects examined the effects of ritonavir (100 mg twice daily) on fluticasone nasal spray (200 mcg daily). In most subjects, fluticasone was undetectable (<10 pg/ml) when administered alone. In subjects in whom fluticasone was detectable when given alone, Cmax and area-under-curve AUC averaged 11.9 pg/ml and 8.43 pg x hr/ml, respectively. With concurrent ritonavir, fluticasone Cmax and AUC increased to 318 pg/ml and 3102.6 pg x hr/ml, respectively.(6-8) This reflects increases in Cmax and AUC by 25-fold and 350-fold, respectively.(6) The cortisol AUC decreased by 86%.(10-13) In a study in 10 healthy subjects, ritonavir (200 mg twice daily for 4 and 14 days) increased the AUC of a single dose of prednisolone by 1.41-fold and 1.30-fold, respectively, after 4 days and 14 days of ritonavir.(14) There have been several case reports of Cushing's syndrome in patients treated concurrently with ritonavir and inhaled budesonide,(15-16) dexamethasone,(17) injectable triamcinolone,(18-21) nasal fluticasone.(23-41) Hepatitis has also been reported with concurrent budesonide and ritonavir.(42) In a study in 9 healthy subjects, mibefradil (50 mg once daily for 3 days) increased the AUC, Cmax, and elimination half-life of methylprednisolone by 3.8-fold, 1.8-fold, and 2.7-fold, respectively.(43) In a study in 8 healthy subjects, following nefazodone administration the following changes were seen with methylprednisolone: mean (+/-SD) area under the concentration-time curve was significantly higher (1393 +/- 343 vs. 2966 +/- 928 ug*h/L; P < 0.005), apparent clearance was lower (28.7 +/- 7.2 vs. 14.6 +/- 7.8 L/h; P < 0.02) and the terminal elimination half-life was longer (2.28 +/- 0.49 vs. 3.32 +/- 0.95 hours; P < 0.02).(44) Selected steroids metabolized by CYP3A4 are linked to this monograph.(45) Selected CYP3A4 inhibitors linked to this monograph include: adagrasib, boceprevir, ceritinib, clarithromycin, erythromycin, itraconazole, josamycin, ketoconazole, lonafarnib, mibefradil, nefazodone, paritaprevir, posaconazole, ribociclib, telaprevir, telithromycin, troleandomycin, tucatinib, and voriconazole.(1-2,45) |
CLARITHROMYCIN, CLARITHROMYCIN ER, E.E.S. 200, E.E.S. 400, ERY-TAB, ERYPED 200, ERYPED 400, ERYTHROCIN LACTOBIONATE, ERYTHROCIN STEARATE, ERYTHROMYCIN, ERYTHROMYCIN ESTOLATE, ERYTHROMYCIN ETHYLSUCCINATE, ERYTHROMYCIN LACTOBIONATE, ITRACONAZOLE, ITRACONAZOLE MICRONIZED, KETOCONAZOLE, KISQALI, KRAZATI, LANSOPRAZOL-AMOXICIL-CLARITHRO, NEFAZODONE HCL, NOXAFIL, OMECLAMOX-PAK, POSACONAZOLE, SPORANOX, TOLSURA, TUKYSA, VFEND, VFEND IV, VOQUEZNA TRIPLE PAK, VORICONAZOLE, VORICONAZOLE (HPBCD), ZOKINVY, ZYKADIA |
| Glucagon (Diagnostic)/Anticholinergics SEVERITY LEVEL: 2-Severe Interaction: Action is required to reduce the risk of severe adverse interaction. MECHANISM OF ACTION: Glucagon and anticholinergic agents may have additive effects on inhibition of gastrointestinal motility.(1) CLINICAL EFFECTS: Concurrent use of glucagon with anticholinergic agents may increase the risk of gastrointestinal hypomotility, including constipation and bowel complications.(1) PREDISPOSING FACTORS: None determined. PATIENT MANAGEMENT: Concurrent use of glucagon as a diagnotic aid is not recommended with the use of anticholinergic agents.(1) If concurrent use is necessary, evaluate the patient's bowel function. Monitor for symptoms of constipation and gastrointestinal hypomotility. DISCUSSION: Both glucagon and anticholinergic agents may have additive effects on inhibition of gastrointestinal motility and increase the risk of gastrointestinal adverse effects.(1) |
GLUCAGON HCL |
| Sodium Iodide I 131/Agents that Affect Iodide SEVERITY LEVEL: 2-Severe Interaction: Action is required to reduce the risk of severe adverse interaction. MECHANISM OF ACTION: Many compounds can affect iodide protein binding and alter iodide pharmacokinetics and pharmacodynamics.(1,2) CLINICAL EFFECTS: Compounds that affect iodide pharmacokinetics and pharmacodynamics may impact the effectiveness of radioactive iodide.(1,2) PREDISPOSING FACTORS: Compounds that affect iodide pharmacokinetics and pharmacodynamics are expected to have the most impact during therapy using radioactive iodide. Diagnostic procedures would be expected to be impacted less. PATIENT MANAGEMENT: Discuss the use of agents that affect iodide pharmacokinetics and pharmacodynamics with the patient's oncologist.(1,2) Because indocyanine green contains sodium iodide, the iodine-binding capacity of thyroid tissue may be reduced for at least one week following administration. Do not perform radioactive iodine uptake studies for at least one week following administration of indocyanine green.(3) The manufacturer of iopamidol states administration may interfere with thyroid uptake of radioactive iodine and decrease therapeutic and diagnostic efficacy. Avoid thyroid therapy or testing for up to 6 weeks post administration of iopamidol.(4) DISCUSSION: Many agents interact with radioactive iodine. The average duration of effect is: anticoagulants - 1 week antihistamines - 1 week anti-thyroid drugs, e.g: carbimazole, methimazole, propylthiouracil - 3-5 days corticosteroids - 1 week iodide-containing medications, e.g: amiodarone - 1-6 months expectorants - 2 weeks Lugol solution - 3 weeks saturated solution of potassium iodine - 3 weeks vitamins - 10-14 days iodide-containing X-ray contrast agents - up to 1 year lithium - 4 weeks phenylbutazone - 1-2 weeks sulfonamides - 1 week thyroid hormones (natural or synthetic), e.g.: thyroxine - 4 weeks tri-iodothyronine - 2 weeks tolbutamide - 1 week topical iodide - 1-9 months (1,2) |
HICON, SODIUM IODIDE I-131 |
There are 8 moderate interactions.
The clinician should assess the patient’s characteristics and take action as needed. Actions required for moderate interactions include, but are not limited to, discontinuing one or both agents, adjusting dosage, altering administration.
| Drug Interaction | Drug Names |
|---|---|
| Tamoxifen/Selected Weak CYP2D6 Inhibitors SEVERITY LEVEL: 3-Moderate Interaction: Assess the risk to the patient and take action as needed. MECHANISM OF ACTION: Inhibitors of CYP2D6 may inhibit the conversion of tamoxifen to endoxifen (an active metabolite of tamoxifen).(1-2) The role of endoxifen in tamoxifen's efficacy has been debated and may involve a minimum concentration level.(3-5) CLINICAL EFFECTS: Concurrent use of inhibitors of CYP2D6 may decrease the effectiveness of tamoxifen in preventing breast cancer recurrence. PREDISPOSING FACTORS: Concurrent use of weak CYP2D6 inhibitors in patients who are CYP2D6 intermediate metabolizers should be avoided. Patients who are CYP2D6 poor metabolizers lack CYP2D6 function and are not affected by CYP2D6 inhibition. PATIENT MANAGEMENT: Although data on this interaction are conflicting, it may be prudent to use alternatives to CYP2D6 inhibitors when possible in patients taking tamoxifen. The US manufacturer of tamoxifen states that the impact on the efficacy of tamoxifen by strong CYP2D6 inhibitors is uncertain and makes no recommendation regarding coadministration with inhibitors of CYP2D6.(12) The manufacturer of paroxetine (a strong CYP2D6 inhibitor) states that alternative agents with little or no CYP2D6 inhibition should be considered.(13) The National Comprehensive Cancer Network's breast cancer guidelines advises caution when coadministering strong CYP2D6 inhibitors with tamoxifen.(14) If concurrent therapy is warranted, the risks versus benefits should be discussed with the patient. DISCUSSION: Some studies have suggested that administration of fluoxetine, paroxetine, and quinidine with tamoxifen or a CYP2D6 poor metabolizer phenotype may result in a decrease in the formation of endoxifen (an active metabolite of tamoxifen) and a shorter time to breast cancer recurrence.(1-2,9) A retrospective study of 630 breast cancer patients found an increasing risk of breast cancer mortality with increasing durations of coadministration of tamoxifen and paroxetine. In the adjusted analysis, absolute increases of 25%, 50%, and 75% in the proportion of time of overlapping use of tamoxifen with paroxetine was associated with 24%, 54%, and 91% increase in the risk of death from breast cancer, respectively.(16) The CYP2D6 genotype of the patient may have a role in the effects of this interaction. Patients with wild-type CYP2D6 genotype may be affected to a greater extent by this interaction. Patients with a variant CYP2D6 genotype may have lower baseline levels of endoxifen and may be affected to a lesser extent by this interaction.(6-10) In a retrospective review, 1,325 patients treated with tamoxifen for breast cancer were classified as being poor 2D6 metabolizers (lacking functional CYP2D6 enzymes), intermediate metabolizers (heterozygous alleles), or extensive metabolizers (possessing 2 functional alleles). After a mean follow-up period of 6.3 years, the recurrence rates were 14.9%, 20.9%, and 29.0%, in extensive metabolizers, intermediate metabolizers, and poor metabolizers, respectively.(11) In October of 2006, the Advisory Committee Pharmaceutical Science, Clinical Pharmacology Subcommittee of the US Food and Drug Administration recommended that the US tamoxifen labeling be updated to include information about the increased risk of breast cancer recurrence in poor CYP2D6 metabolizers (either by genotype or drug interaction).(17-18) The labeling changes were never made due to ongoing uncertainty about the effects of CYP2D6 genotypes on tamoxifen efficacy. In contrast to the above information, two studies have shown no relationship between CYP2D6 genotype and breast cancer outcome.(19-21) As well, a number of studies found no association between use of CYP2D6 inhibitors and/or antidepressants in patients on tamoxifen and breast cancer recurrence,(22-26) though the studies were limited by problematic selection of CYP2D6 inhibitors and short follow-up. Weak inhibitors of CYP2D6 include: alogliptin, artesunate, celecoxib, chloroquine, cimetidine, clobazam, clomipramine, cobicistat, delavirdine, diltiazem, dimenhydrinate, diphenhydramine, dronabinol, dupilumab, echinacea, enasidenib, fedratinib, felodipine, fluvoxamine, gefitinib, hydralazine, imatinib, labetalol, lorcaserin, nicardipine, osilodrostat, ranitidine, ritonavir, sertraline, verapamil and viloxazine.(27) |
SOLTAMOX, TAMOXIFEN CITRATE |
| Eliglustat/Weak CYP2D6 Inhibitors SEVERITY LEVEL: 3-Moderate Interaction: Assess the risk to the patient and take action as needed. MECHANISM OF ACTION: Weak inhibitors of CYP2D6 may inhibit the metabolism of eliglustat. If the patient is also taking an inhibitor of CYP3A4, eliglustat metabolism can be further inhibited.(1) CLINICAL EFFECTS: Concurrent use of an agent that is a weak inhibitor of CYP2D6 may result in elevated levels of and clinical effects of eliglustat, including prolongation of the PR, QTc, and/or QRS intervals, which may result in life-threatening cardiac arrhythmias.(1) PREDISPOSING FACTORS: If the patient is also taking an inhibitor of CYP3A4 and/or has hepatic impairment, eliglustat metabolism can be further inhibited.(1) The risk of QT prolongation or torsades de pointes may be increased in patients with cardiovascular disease (e.g. heart failure, myocardial infarction, history of torsades de pointes, congenital long QT syndrome), hypokalemia, hypomagnesemia, hypocalcemia, bradycardia, female gender, or advanced age.(2) Concurrent use of more than one drug known to cause QT prolongation or higher systemic concentrations of either QT prolonging drug are additional risk factors for torsades de pointes. Factors which may increase systemic drug concentrations include rapid infusion of an intravenous dose or impaired metabolism or elimination of the drug (e.g. coadministration with an agent which inhibits its metabolism or elimination, genetic impairment in drug metabolism or elimination, and/or renal/hepatic dysfunction).(2) PATIENT MANAGEMENT: The dosage of eliglustat with weak inhibitors of CYP2D6 in poor CYP2D6 metabolizers should be limited to 84 mg daily.(1) The dosage of eliglustat with weak inhibitors of CYP2D6 in extensive CYP2D6 metabolizers with mild (Child-Pugh Class A) hepatic impairment should be limited to 84 mg daily.(1) If concurrent therapy is warranted, consider obtaining serum calcium, magnesium, and potassium levels and monitoring ECG at baseline and at regular intervals. Correct any electrolyte abnormalities. Instruct patients to report any irregular heartbeat, dizziness, or fainting. DISCUSSION: Paroxetine (30 mg daily), a strong inhibitor of CYP2D6, increased eliglustat (84 mg BID) maximum concentration (Cmax) and area-under-curve (AUC) by 7-fold and 8.4-fold, respectively, in extensive metabolizers. Physiologically-based pharmacokinetic (PKPB) models suggested paroxetine would increase eliglustat Cmax and AUC by 2.1-fold and 2.3-fold, respectively, in intermediate metabolizers. PKPB models suggested ketoconazole may increase the Cmax and AUC of eliglustat (84 mg daily) by 4.3-fold and 6.2-fold, respectively, in poor metabolizers.(1) PKPB models suggested terbinafine, a moderate inhibitor of CYP2D6, would increase eliglustat Cmax and AUC by 3.8-fold and 4.5-fold, respectively, in extensive metabolizers and by 1.6-fold and 1.6-fold, respectively in intermediate metabolizers. PKPB models suggest that concurrent eliglustat (84 mg BID), paroxetine (a strong inhibitor of CYP2D6), and ketoconazole would increase eliglustat Cmax and AUC by 16.7-fold and 24.2-fold, respectively, in extensive metabolizers. In intermediate metabolizers, eliglustat Cmax and AUC would be expected to increase 7.5-fold and 9.8-fold, respectively.(1) PKPB models suggest that concurrent eliglustat (84 mg BID), terbinafine (a moderate inhibitor of CYP2D6), and ketoconazole would increase eliglustat Cmax and AUC by 10.2-fold and 13.6-fold, respectively, in extensive metabolizers. In intermediate metabolizers, eliglustat Cmax and AUC would be expected to increase 4.2-fold and 5-fold, respectively.(1) A single dose of rolapitant increased dextromethorphan, a CYP2D6 substrate, about 3-fold on days 8 and day 22 following administration. Dextromethorphan levels remained elevated by 2.3-fold on day 28 after single dose rolapitant. The inhibitory effects of rolapitant on CYP2D6 are expected to persist beyond 28 days.(5) Weak inhibitors of CYP2D6 include: alogliptin, artesunate, celecoxib, chloroquine, clobazam, clomipramine, desvenlafaxine, dimenhydrinate, diphenhydramine, dronabinol, dupilumab, echinacea, enasidenib, felodipine, gefitinib, hydralazine, hydroxychloroquine, lorcaserin, methadone, panobinostat, propafenone, sertraline, vemurafenib, and venlafaxine.(3,4) |
CERDELGA |
| Metoprolol/Selected CYP2D6 Inhibitors SEVERITY LEVEL: 3-Moderate Interaction: Assess the risk to the patient and take action as needed. MECHANISM OF ACTION: CYP2D6 inhibitors may inhibit the metabolism of metoprolol.(1,2) CLINICAL EFFECTS: Concurrent use of CYP2D6 inhibitors may result in elevated levels of and toxicity from metoprolol.(1,2) PREDISPOSING FACTORS: The interaction may be more severe in patients who are ultrarapid metabolizers of CYP2D6,(1,2) elderly,(3) and on higher doses of beta-blockers.(3) PATIENT MANAGEMENT: Monitor patients receiving concurrent therapy with metoprolol and inhibitors of CYP2D6. The dosage of metoprolol may need to be adjusted.(1,2) The effects of rolapitant, a moderate CYP2D6 inhibitor, on CYP2D6 are expected to last at least 28 days after administration.(4) DISCUSSION: In a case report, a patient maintained on metoprolol developed bradycardia following the addition of bupropion.(5) In a study in 20 healthy females, diphenhydramine increased the AUC of metoprolol by 21%. Heart rate reduction increased 29%.(6) In a randomized study in 16 healthy subjects, diphenhydramine decreased metoprolol oral and nonrenal clearance by 2-fold in extensive 2D6 metabolizers. In extensive 2D6 metabolizers, metoprolol-induced effects on heart rate, systolic blood pressure, and aortic blood flow peak velocity were all increased. There were no effects of diphenhydramine in poor metabolizers.(7) Fluoxetine has been shown to inhibit metoprolol metabolism in vitro.(8) There is a case report of severe bradycardia following the addition of fluoxetine to metoprolol.(9) In a 3-way, randomized, cross-over study in healthy subjects, paroxetine (20 mg daily) increased the area-under-curve (AUC) of both S- and R-metoprolol by 3-fold, and 4-fold, respectively, regardless of whether the formulation of metoprolol was immediate release or extended release. Concurrent paroxetine also significantly decreased heart rate and blood pressure when compared to metoprolol alone.(10) In an open-label, randomized, cross-over study in 10 healthy subjects, paroxetine increased the AUC of S-metoprolol and R-metoprolol from an immediate release formulation (50 mg)by 4-fold and 5-fold, respectively. Paroxetine increased the AUC of S-metoprolol and R-metoprolol from an extended release formulation (100 mg) by 3-fold and 4-fold, respectively.(11) In a study in patients with acute myocardial infarction and depression, paroxetine (20 mg daily) increased the AUC of metoprolol 3-fold. Mean heart rate was significantly lower following the addition of paroxetine to metoprolol. Two patients experienced bradycardia and severe orthostatic hypotension.(12) In an open trial in 8 healthy males, paroxetine (20 mg daily) increased the AUC of S-metoprolol and R-metoprolol by 4-fold and 7-fold, respectively.(13) There are case reports of complete atrioventricular block(14) and bradycardia(15) with concurrent metoprolol and paroxetine. A systematic review and meta-analysis of CYP2D6 interactions between metoprolol and either paroxetine or fluoxetine reviewed 9 articles including 4 primary and 2 observational studies as well as 3 case reports. Experimental studies noted paroxetine increased the AUC of metoprolol 3-fold to 5-fold and significantly decreased blood pressure and heart rate. Paroxetine and fluoxetine have shown equipotent inhibitor capacity on CYP2D6. The metabolite, norfluoxetine, is also an inhibitor of CYP2D6.(16) A retrospective cohort study evaluated morbidity in patients on a beta-blocker primarily metabolized by CYP2D6 (e.g., nebivolol, metoprolol, carvedilol, propranolol, labetalol) and started on a strong or moderate CYP2D6-inhibiting antidepressant (e.g., fluoxetine, paroxetine, bupropion, duloxetine). Use of such an antidepressant with a beta-blocker was associated with an increased risk of hospitalization or ED visit due to an adverse hemodynamic event (HR 1.53, 95% CI 1.03-2.81, p=0.04).(3) CYP2D6 inhibitors include: abiraterone, bupropion, celecoxib, cinacalcet, citalopram, dacomitinib, dimenhydrinate, diphenhydramine, duloxetine, escitalopram, fedratinib, fluoxetine, hydroxychloroquine, imatinib, lorcaserin, osilodrostat, paroxetine, ranitidine, ranolazine, rolapitant, and sertraline. One or more of the drug pairs linked to this monograph have been included in a list of interactions that could be considered for classification as "non-interruptive" in EHR systems. This DDI subset was vetted by an expert panel commissioned by the U.S. Office of the National Coordinator (ONC) for Health Information Technology. |
KAPSPARGO SPRINKLE, LOPRESSOR, METOPROLOL SUCCINATE, METOPROLOL TARTRATE, METOPROLOL-HYDROCHLOROTHIAZIDE, TOPROL XL |
| Zonisamide/Anticholinergics SEVERITY LEVEL: 3-Moderate Interaction: Assess the risk to the patient and take action as needed. MECHANISM OF ACTION: Zonisamide can cause decreased sweating and elevated body temperature. Agents with anticholinergic activity can predispose patients to heat-related disorders.(1-2) CLINICAL EFFECTS: Concurrent use of zonisamide with agents with anticholinergic activity may increase the incidence of oligohidrosis and hyperthermia, especially in pediatric or adolescent patients.(1-2) Overheating and dehydration can lead to brain damage and death. PREDISPOSING FACTORS: Pediatric and adolescent patients and patients with dehydration may be more likely to experience heat-related disorders.(1) PATIENT MANAGEMENT: The UK and US manufacturers of zonisamide state that caution should be used in adults when zonisamide is prescribed with other medicinal products that predispose to heat-related disorders, such as agents with anticholinergic activity.(1-2) Pediatric and adolescent patients must not take anticholinergic agents (e.g. clomipramine, hydroxyzine, diphenhydramine, haloperidol, imipramine, and oxybutynin) concurrently with zonisamide.(1) Monitor for signs and symptoms of heat stroke: skin feels very hot with little or no sweating, confusion, muscle cramps, rapid heartbeat, or rapid breathing. Monitor for signs and symptoms of dehydration: dry mouth, urinating less than usual, dark-colored urine, dry skin, feeling tired, dizziness, or irritability. If signs or symptoms of dehydration, oligohidrosis, or elevated body temperature occur, discontinuation of zonisamide should be considered. DISCUSSION: Case reports of decreased sweating and elevated temperature have been reported, especially in pediatric patients. Some cases resulted in heat stroke that required hospital treatment and resulted in death.(1) |
ZONEGRAN, ZONISADE, ZONISAMIDE |
| Topiramate/Anticholinergics SEVERITY LEVEL: 3-Moderate Interaction: Assess the risk to the patient and take action as needed. MECHANISM OF ACTION: Topiramate can cause decreased sweating and elevated body temperature. Agents with anticholinergic activity can predispose patients to heat-related disorders.(1-2) CLINICAL EFFECTS: Concurrent use of topiramate with agents with anticholinergic activity may increase the incidence of oligohidrosis and hyperthermia, especially in pediatric or adolescent patients.(1-2) Overheating and dehydration can lead to brain damage and death. PREDISPOSING FACTORS: Pediatric and adolescent patients and patients with dehydration may be more likely to experience heat-related disorders.(1) PATIENT MANAGEMENT: The manufacturer of topiramate states that caution should be used when topiramate is prescribed with other medicinal products that predispose to heat-related disorders, such as agents with anticholinergic activity (e.g. clomipramine, hydroxyzine, diphenhydramine, haloperidol, imipramine, and oxybutynin) concurrently with zonisamide.(1) Monitor for signs and symptoms of heat stroke: skin feels very hot with little or no sweating, confusion, muscle cramps, rapid heartbeat, or rapid breathing. Monitor for signs and symptoms of dehydration: dry mouth, urinating less than usual, dark-colored urine, dry skin, feeling tired, dizziness, or irritability. If signs or symptoms of dehydration, oligohidrosis, or elevated body temperature occur, discontinuation of zonisamide should be considered. DISCUSSION: Case reports of decreased sweating and elevated temperature have been reported, especially in pediatric patients. Some cases resulted in heat stroke that required hospital treatment.(1) A 64-year old woman developed non-exertional hyperthemia while taking multiple psychiatric medications with topiramate.(2) |
EPRONTIA, PHENTERMINE-TOPIRAMATE ER, QSYMIA, TOPAMAX, TOPIRAMATE, TOPIRAMATE ER, TOPIRAMATE ER SPRINKLE, TROKENDI XR |
| Selected Corticosteroids/Levoketoconazole SEVERITY LEVEL: 3-Moderate Interaction: Assess the risk to the patient and take action as needed. MECHANISM OF ACTION: Levoketoconazole may inhibit the CYP3A4 mediated metabolism of some corticosteroids, resulting in increased systemic exposure. Levoketoconazole may also suppress endogenous cortisol output. Levoketoconazole is the enantiomer of ketoconazole. CLINICAL EFFECTS: Concurrent use of levoketoconazole may result in elevated levels of and effects from the corticosteroid, including Cushing syndrome. These effects have been seen with systemic as well as inhaled corticosteroids. PREDISPOSING FACTORS: None determined. PATIENT MANAGEMENT: Patients should be carefully monitored with concurrent administration of these agents, or when levoketoconazole is added to corticosteroid therapy. The dose of the corticosteroid may need to be adjusted or alternative therapy considered. DISCUSSION: The concurrent use of ketoconazole has been shown to increase budesonide area-under-curve (AUC) by eight-fold. In a study in eight healthy subjects, the simultaneous administration of ketoconazole increased budesonide AUC by 6.5-fold. Administering the two agents 12 hours apart increased budesonide AUC by 3.8-fold. In a study in 6 healthy subjects, pretreatment with ketoconazole (200 mg daily) increased the AUC of a single intravenous dose of methylprednisolone (20 mg) by 135% and decreased its clearance by 60%. Concurrent ketoconazole also increased the reduction in 24-hour cortisol AUC and suppressed morning cortisol concentrations. In a study in 8 healthy subjects, ketoconazole decreased the clearance of methylprednisolone by 46% and increased mean residence time by 37%. In a randomized, cross-over study in 6 healthy subjects, pretreatment with ketoconazole (200 mg daily for 6 days) had no effect on the pharmacokinetics of a single intravenous dose of prednisolone (14.8 mg). In a study, concurrent oral ketoconazole increased the AUC of des-ciclesonide from orally inhaled ciclesonide by 3.6-fold. There were no changes in ciclesonide levels. In a study in 24 healthy subjects, subjects were randomized to receive either ketoconazole (200 mg BID) or placebo on Days 4-9 of a a 9 day course of mometasone (400 mcg BID). No subject had mometasone levels greater than 150 pcg/ml on Day 3. Four of 12 subjects who received ketoconazole had mometasone Cmax levels greater than 200 mcg/ml on Day 9. Plasma cortisol levels appeared to decrease as well. In a cross-over study in 15 healthy subjects, subjects were randomized to receive fluticasone furoate and vilanterol on days 5-11 with either ketoconazole (200mg once daily) or placebo for days 1-11 with a washout period of 7-14 days. Fluticasone furoate AUC was increased by 36%, Cmax was increased by 33%, and decreased systemic cortisol levels by 27%. There were no effects on heart rate and blood potassium levels. There was a small increase in QTc which was 7.6ms greater when compared to placebo; however, ketoconazole has been reported to increase QTc by 5-6ms. Vilanterol AUC was increased by 65% and Cmax was increased by 22%. There were no effects on heart rate and blood potassium levels. No serious adverse events occurred and no subjects withdrew from the study due to adverse events. The most common adverse event reported was headache. Coadministration of orally inhaled fluticasone (1000 mcg) and ketoconazole (200 mg once daily) resulted in a 1.9-fold increase in plasma fluticasone exposure and a 45% decrease in plasma cortisol AUC. |
RECORLEV |
| Selected Steroids/Nirmatrelvir-Ritonavir SEVERITY LEVEL: 3-Moderate Interaction: Assess the risk to the patient and take action as needed. MECHANISM OF ACTION: Nirmatrelvir/ritonavir may inhibit the metabolism of corticosteroids metabolized by CYP3A4.(1) CLINICAL EFFECTS: Nirmatrelvir/ritonavir may result in increased systemic exposure to and effects from corticosteroids metabolized by CYP3A4.(1) PREDISPOSING FACTORS: None determined. PATIENT MANAGEMENT: Coadministration with corticosteroids of all routes of administration of which exposures are significantly increased by strong CYP3A4 inhibitors can increase the risk of Cushing's syndrome and adrenal suppression. However, the risk of Cushing's syndrome and adrenal suppression associated with short-term use of a strong CYP3A4 inhibitor is low.(1) The manufacturer of nirmatrelvir/ritonavir recommends considering alternative corticosteroids including beclomethasone, prednisone, and prednisolone.(1) DISCUSSION: Concurrent use of a single dose of midazolam 2 mg, a CYP3A4 substrate, with nirmatrelvir-ritonavir (300 mg/100 mg twice daily for nine doses) increased the maximum concentration (Cmax) and area-under-curve (AUC) of midazolam by 37% and 143%, respectively.(1) A study in 18 healthy subjects examined the effects of ritonavir (100 mg twice daily) on fluticasone nasal spray (200 mcg daily). In most subjects, fluticasone was undetectable (<10 pg/ml) when administered alone. In subjects in whom fluticasone was detectable when given alone, Cmax and area-under-curve AUC averaged 11.9 pg/ml and 8.43 pg x hr/ml, respectively. With concurrent ritonavir, fluticasone Cmax and AUC increased to 318 pg/ml and 3102.6 pg x hr/ml, respectively.(7,11,13) This reflects increases in Cmax and AUC by 25-fold and 350-fold, respectively.(3) The cortisol AUC decreased by 86%.(3-6) In a study in 10 healthy subjects, ritonavir (200 mg twice daily for 4 and 14 days) increased the AUC of a single dose of prednisolone by 1.41-fold and 1.30-fold, respectively, after 4 days and 14 days of ritonavir.(7) Selected steroids linked to this monograph include: budesonide, ciclesonide, dexamethasone, fluticasone, methylprednisolone, and triamcinolone.(8) |
PAXLOVID |
| Propranolol/Selected CYP2D6 Inhibitors SEVERITY LEVEL: 3-Moderate Interaction: Assess the risk to the patient and take action as needed. MECHANISM OF ACTION: CYP2D6 inhibitors may inhibit the metabolism of propranolol.(1) CLINICAL EFFECTS: Concurrent use of CYP2D6 inhibitors may result in elevated levels of and toxicity from propranolol, including hypotension and bradycardia.(1) PREDISPOSING FACTORS: None determined. PATIENT MANAGEMENT: Monitor patients receiving concurrent therapy with propranolol and CYP2D6 inhibitors. The dosage of propranolol may need to be adjusted.(1) DISCUSSION: In a pharmacokinetic study in 16 healthy volunteers, concurrent use of quinidine 200 mg (a CYP2D6 inhibitor) increased the area-under-curve (AUC) of propranolol by 2.29-fold.(2) In a pharmacokinetic study in 6 healthy subjects, concurrent use of quinidine increased propranolol AUC 2-fold.(3) A retrospective review of concurrent use of propranolol and antidepressants evaluated the risk of hospitalization or emergency room visit within 30 days of concurrent prescription. In patients receiving antidepressants with moderate to strong CYP2D6 inhibitory effects, patient were an increased risk compared to patients receiving no antidepressants (Hazard Ratio (HR) = 1.53; 95% CI 1.03-2.81 vs. HR = 1.24; 95% CI 0.82-1.88).(4) Case reports of bradycardia and cardiac adverse effects have been reported with concurrent use of propranolol and the antidepressants fluoxetine and paroxetine (strong CYP2D6 inhibitors).(5) Strong CYP2D6 inhibitors include: bupropion, dacomitinib, fluoxetine, mavorixafor, and paroxetine. Moderate CYP2D6 inhibitors include: abiraterone, asunaprevir, berotralstat, capivasertib, cinacalcet, duloxetine, eliglustat, escitalopram, lorcaserin, mirabegron, moclobemide, quinine, ranolazine, and rolapitant. Weak CYP2D6 inhibitors include: desvenlafaxine, diphenhydramine, dimenhydrinate, dronabinol, fedratinib, hydroxychloroquine, imatinib, osilodrostat, ranitidine, and sertraline.(6) |
HEMANGEOL, INDERAL LA, INDERAL XL, INNOPRAN XL, PROPRANOLOL HCL, PROPRANOLOL HCL ER |
The following contraindication information is available for ALLERGY RELIEF (cetirizine hcl):
Drug contraindication overview.
No enhanced Contraindications information available for this drug.
No enhanced Contraindications information available for this drug.
There are 0 contraindications.
There are 16 severe contraindications.
Adequate patient monitoring is recommended for safer drug use.
| Severe List |
|---|
| Angle-closure glaucoma |
| Benign prostatic hyperplasia |
| Bladder outflow obstruction |
| Chronic idiopathic constipation |
| Chronic kidney disease stage 4 (severe) GFR 15-29 ml/min |
| Chronic kidney disease stage 5 (failure) GFr<15 ml/min |
| Nasal septal perforation |
| Nasal septal ulcers |
| Nasal trauma |
| Ocular herpes simplex |
| Ocular hypertension |
| Open angle glaucoma |
| Operation on nose |
| Severe infection |
| Stenosing peptic ulcer |
| Urinary retention |
There are 17 moderate contraindications.
Clinically significant contraindication, where the condition can be managed or treated before the drug may be given safely.
| Moderate List |
|---|
| Adrenocortical insufficiency |
| Cataracts |
| Coronary artery disease |
| Disease of liver |
| Epistaxis |
| Glaucoma |
| Hepatic failure |
| Hypertension |
| Hyperthyroidism |
| Inactive tuberculosis |
| Kidney disease with reduction in glomerular filtration rate (GFr) |
| Ocular hypertension |
| Parasitic infection |
| Pyloroduodenal obstruction |
| Renal disease with moderate to severe function impairment |
| Seizure disorder |
| Urinary retention |
The following adverse reaction information is available for ALLERGY RELIEF (cetirizine hcl):
Adverse reaction overview.
No enhanced Common Adverse Effects information available for this drug.
No enhanced Common Adverse Effects information available for this drug.
There are 40 severe adverse reactions.
| More Frequent | Less Frequent |
|---|---|
| None. |
Bronchitis Lesion of nasal mucosa Urticaria |
| Rare/Very Rare |
|---|
|
Abnormal hepatic function tests Acute bacterial otitis media Acute generalized exanthematous pustulosis Adrenocortical insufficiency Anaphylaxis Angioedema Blood dyscrasias Bronchospastic pulmonary disease Cardiac arrhythmia Cataracts Cholestasis Contact dermatitis Dehydration Edema Extrasystoles Glaucoma Glomerulonephritis Hallucinations Hearing loss Hemolytic anemia Hepatitis Hypercortisolism Hypersensitivity drug reaction Hypotension Immunosuppression Impaired wound healing Nasal candidiasis Nasal septal perforation Palpitations Pharyngeal candidiasis Seizure disorder Skin rash Thrombocytopenic disorder Tongue swelling Tonic clonic seizure Upper respiratory infection Urticaria |
There are 127 less severe adverse reactions.
| More Frequent | Less Frequent |
|---|---|
|
Anticholinergic toxicity Dizziness Drowsy Dry nose Dry throat Epistaxis Headache disorder Nasal passage irritation Pharyngitis Thick bronchial secretions Xerostomia |
Acute abdominal pain Back pain Conjunctivitis Cough Crusted nasal mucosa Diarrhea Dizziness Drowsy Dysgeusia Dysmenorrhea Dyspepsia Epistaxis Erythema of nasal mucosa Fatigue Fever Headache disorder Insomnia Muscle weakness Myalgia Nasal congestion Nasal pain Nausea Pain in oropharynx Pharyngitis Rhinorrhea Sedation Sinusitis Skin rash Sore throat Viral infection Vomiting Xerostomia |
| Rare/Very Rare |
|---|
|
Abdominal distension Abdominal pain with cramps Accidental fall Acne vulgaris Acute abdominal pain Acute cognitive impairment Aggressive behavior Agitation Alopecia Anorexia Anticholinergic toxicity Ataxia Blurred vision Bronchitis Chest discomfort Chills Concentration difficulty Constipation Cough Cramps in legs Diarrhea Diplopia Dizziness Dream disorder Dry nose Dry throat Dysgeusia Dyskinesia Dyspepsia Dyspnea Dysuria Earache Epistaxis Eructation Euphoria Excitement Fatigue Fever Flatulence Flu-like symptoms Gastritis Hallucinations Headache disorder Hemorrhoids Hyperhidrosis Hyperkinesis Increased appetite Insomnia Irritability Maculopapular rash Malaise Memory impairment Migraine Nausea Nervousness Nightmares Pain Palpitations Paresthesia Parosmia Pharyngitis Pruritus of skin Sedation Skin photosensitivity Skin rash Stomatitis Symptoms of anxiety Syncope Tachycardia Tinnitus Tongue discoloration Toothache Tremor Unsteady gait Urinary incontinence Urinary retention Urticaria Vertigo Visual changes Voice change Vomiting Weight gain Wheezing Xerostomia |
The following precautions are available for ALLERGY RELIEF (cetirizine hcl):
No enhanced Pediatric Use information available for this drug.
Contraindicated
Severe Precaution
Management or Monitoring Precaution
Contraindicated
| None |
Severe Precaution
| None |
Management or Monitoring Precaution
| None |
Fluticasone propionate nasal spray should be used during pregnancy only when the potential benefits justify the possible risks to the fetus. Since adrenal insufficiency may occur in neonates born to women who received corticosteroids during pregnancy, these neonates should be monitored carefully for manifestations of this condition. Although there are no adequate and well-controlled studies to date using fluticasone propionate in pregnant women, fluticasone propionate has been shown to be teratogenic and embryotoxic in mice or rats when administered subcutaneously at daily dosages of 45 or 100 mcg/kg, respectively (approximately equivalent to 4 times the maximum recommended daily intranasal dosage in adults based on surface area).
Observed fetal toxicity was characteristic of potent glucocorticoid compounds and included embryonic growth retardation, omphalocele, cleft palate, and retarded cranial ossification. Fetal weight reduction and cleft palate were observed in offspring of rabbits given fluticasone propionate 4 mcg/kg subcutaneously (less than the maximum recommended daily intranasal dosage in adults). Following oral administration of up to 300 mcg/kg (approximately 25 times the maximum recommended daily intranasal dosage in adults on a mcg/m2 basis) daily of fluticasone propionate in rabbits, the drug was not detected in plasma, and no maternal effects nor increased incidence of external, visceral, or skeletal fetal defects was detected.
The low bioavailability and small distribution of fluticasone propionate across the placenta may account for the lack of teratogenicity; in rats and rabbits receiving oral fluticasone propionate dosages of 100 mcg/kg (590 mcg/m2) or 300 mcg/kg (3.6 mg/m2), respectively, less than 0.008% of the dose crossed the placenta. Long-term experience with the use of oral glucocorticoids suggests that rodents are more prone to teratogenic effects of glucocorticoids than humans.
In addition, because there is a natural increase in glucocorticoid production during pregnancy, most women will require a lower exogenous glucocorticoid dose and many will not need glucocorticoid treatment during pregnancy. Reproduction studies in rats and rabbits receiving diphenhydramine hydrochloride dosages up to 5 times the recommended human dosage have not revealed evidence of harm to the fetus. However, diphenhydramine has been shown to cross the placenta.
In one epidemiologic study, use of bromodiphenhydramine (no longer commercially available) but not diphenhydramine was associated with an increased risk of teratogenic effects. In another epidemiologic study, there also was no evidence of increased risk of teratogenicity associated with diphenhydramine use during the first trimester, although a modest association could not be ruled out. Use of diphenhydramine during the first trimester of pregnancy has been associated with an increased risk of cleft palate alone or combined with other fetal abnormalities, and the drug has been reported to potentiate the teratogenic effect of morphine in mice.
The manufacturers state that there are no adequate and controlled studies to date using diphenhydramine in pregnant women, and the drugs should be used during pregnancy only when clearly needed. Reproduction studies in mice receiving fexofenadine doses up to 3730 mg/kg (approximately 10-15 times the maximum recommended daily oral human dosage of fexofenadine hydrochloride in adults) have not revealed evidence of adverse or teratogenic effects during gestation. Reproduction studies in rats and rabbits using oral terfenadine dosages up to 300 mg/kg resulting in fexofenadine exposure levels calculated to be about 3-4 and 25-31 times, respectively, those resulting from the maximum recommended daily oral human dosage of fexofenadine hydrochloride in adults have not revealed evidence of teratogenicity.
However, in rats, oral terfenadine dosages of 150 mg/kg, resulting in fexofenadine exposure levels calculated to be about 3-4 times those resulting from the maximum recommended daily oral human dosage of fexofenadine hydrochloride in adults (based on comparison of the AUC), were associated with decreased weight gain and neonatal survival in the pups. Reproduction studies in rats and rabbits using terfenadine and pseudoephedrine hydrochloride in a fixed-combination ratio of 1:2 at dosages of 150/300 (corresponding to fexofenadine AUCs of about 3-4 times the maximum recommended adult therapeutic value and to pseudoephedrine hydrochloride dosages about 10 times the maximum recommended human adult daily oral dosage, on a mg/m2 basis) and 100/200 mg/kg daily (corresponding to fexofenadine AUCs of about 8-10 times the maximum recommended adult therapeutic value and to pseudoephedrine hydrochloride dosages about 15 times the maximum recommended human adult daily oral dosage, on a mg/m2 basis), respectively, have revealed evidence of reduced fetal weight; delayed ossification with wavy ribs also was observed in rats receiving the drug at these dosages. There are no adequate and controlled studies to date using fexofenadine in pregnant women, and fexofenadine hydrochloride alone or in fixed combination with pseudoephedrine hydrochloride should be used during pregnancy only when the potential benefits justify the possible risks to the fetus.
An increased incidence of hypospadias in male infants born to women who received loratadine during pregnancy was reported in one study. However, analysis of data from the National Birth Defects Prevention Study (NBDPS) indicated that use of loratadine during early pregnancy was not associated with an increased risk of second- or third-degree hypospadias. In addition, in 2 small prospective cohort studies that surveyed pregnant women who contacted a teratology information service, use of loratadine during the first trimester of pregnancy was not associated with major congenital anomalies and did not affect the rate of live birth, gestational age at birth, and birth weight.
Despite these findings, it should be noted that interpretation of these results is limited by the statistical limitations of the studies (i.e., small sample size, inadequate power, reliance on patient recall of drug use, exclusion criteria). The 2 prospective cohort studies were powered to detect statistical significance only if a substantial (i.e., approximately threefold) increase in the overall rate of major congenital anomalies was observed; the study that relied on NBDPS data excluded first-degree hypospadias because of the difficulty of detecting this mildest form in routine surveillance, making it difficult to determine the relationship between loratadine and this form of hypospadias. Thus, while these data may be useful, further study is needed to completely rule out the teratogenic risk of loratadine.
Because there are no adequate and controlled studies to date using loratadine in pregnant women, loratadine alone or in fixed combination with pseudoephedrine hydrochloride should be used during pregnancy only when the potential benefits justify the possible risks to the fetus. Reproduction studies in rats and rabbits using loratadine dosages up to 75 and 150 times, respectively, the maximum daily human dosage on a mg/m2 basis have not revealed evidence of harm to the fetus. Reproduction studies in mice, rats, and rabbits using oral cetirizine hydrochloride dosages up to 96, 225, and 135 mg/kg daily, respectively (approximately 40, 180, and 220 times, respectively, the maximum recommended daily oral dosage in adults on a mg/m2 basis), have not revealed evidence of teratogenicity.
Because there are no adequate and controlled studies to date using cetirizine in pregnant women and animal studies are not always predictive of human response, cetirizine hydrochloride should be used during pregnancy only when clearly needed. Cetirizine hydrochloride in combination with pseudoephedrine has been shown to increase the number of fetal skeletal malformations (rib distortions) and variants (unossified sternebrae) in rats when given orally in a fixed-combination ratio at a dosage of 6/154 mg/kg (approximately 5 times the maximum recommended adult dosage on a mg/m2 basis). These effects were not observed at a dosage of 1.6/38
mg/kg (approximately the maximum recommended adult dosage on a mg/m2 basis). Reproduction studies in rabbits using cetirizine hydrochloride and pseudoephedrine hydrochloride in a fixed-combination ratio at a dosage of up to 6/154 mg/kg (approximately 10 times the maximum recommended adult dosage on a mg/m2 basis) have not revealed evidence of harm to the fetus. There are no adequate and controlled studies to date using cetirizine hydrochloride and pseudoephedrine hydrochloride in pregnant women, and the fixed combination should be used during pregnancy only when the potential benefits justify the possible risks to the fetus.
Observed fetal toxicity was characteristic of potent glucocorticoid compounds and included embryonic growth retardation, omphalocele, cleft palate, and retarded cranial ossification. Fetal weight reduction and cleft palate were observed in offspring of rabbits given fluticasone propionate 4 mcg/kg subcutaneously (less than the maximum recommended daily intranasal dosage in adults). Following oral administration of up to 300 mcg/kg (approximately 25 times the maximum recommended daily intranasal dosage in adults on a mcg/m2 basis) daily of fluticasone propionate in rabbits, the drug was not detected in plasma, and no maternal effects nor increased incidence of external, visceral, or skeletal fetal defects was detected.
The low bioavailability and small distribution of fluticasone propionate across the placenta may account for the lack of teratogenicity; in rats and rabbits receiving oral fluticasone propionate dosages of 100 mcg/kg (590 mcg/m2) or 300 mcg/kg (3.6 mg/m2), respectively, less than 0.008% of the dose crossed the placenta. Long-term experience with the use of oral glucocorticoids suggests that rodents are more prone to teratogenic effects of glucocorticoids than humans.
In addition, because there is a natural increase in glucocorticoid production during pregnancy, most women will require a lower exogenous glucocorticoid dose and many will not need glucocorticoid treatment during pregnancy. Reproduction studies in rats and rabbits receiving diphenhydramine hydrochloride dosages up to 5 times the recommended human dosage have not revealed evidence of harm to the fetus. However, diphenhydramine has been shown to cross the placenta.
In one epidemiologic study, use of bromodiphenhydramine (no longer commercially available) but not diphenhydramine was associated with an increased risk of teratogenic effects. In another epidemiologic study, there also was no evidence of increased risk of teratogenicity associated with diphenhydramine use during the first trimester, although a modest association could not be ruled out. Use of diphenhydramine during the first trimester of pregnancy has been associated with an increased risk of cleft palate alone or combined with other fetal abnormalities, and the drug has been reported to potentiate the teratogenic effect of morphine in mice.
The manufacturers state that there are no adequate and controlled studies to date using diphenhydramine in pregnant women, and the drugs should be used during pregnancy only when clearly needed. Reproduction studies in mice receiving fexofenadine doses up to 3730 mg/kg (approximately 10-15 times the maximum recommended daily oral human dosage of fexofenadine hydrochloride in adults) have not revealed evidence of adverse or teratogenic effects during gestation. Reproduction studies in rats and rabbits using oral terfenadine dosages up to 300 mg/kg resulting in fexofenadine exposure levels calculated to be about 3-4 and 25-31 times, respectively, those resulting from the maximum recommended daily oral human dosage of fexofenadine hydrochloride in adults have not revealed evidence of teratogenicity.
However, in rats, oral terfenadine dosages of 150 mg/kg, resulting in fexofenadine exposure levels calculated to be about 3-4 times those resulting from the maximum recommended daily oral human dosage of fexofenadine hydrochloride in adults (based on comparison of the AUC), were associated with decreased weight gain and neonatal survival in the pups. Reproduction studies in rats and rabbits using terfenadine and pseudoephedrine hydrochloride in a fixed-combination ratio of 1:2 at dosages of 150/300 (corresponding to fexofenadine AUCs of about 3-4 times the maximum recommended adult therapeutic value and to pseudoephedrine hydrochloride dosages about 10 times the maximum recommended human adult daily oral dosage, on a mg/m2 basis) and 100/200 mg/kg daily (corresponding to fexofenadine AUCs of about 8-10 times the maximum recommended adult therapeutic value and to pseudoephedrine hydrochloride dosages about 15 times the maximum recommended human adult daily oral dosage, on a mg/m2 basis), respectively, have revealed evidence of reduced fetal weight; delayed ossification with wavy ribs also was observed in rats receiving the drug at these dosages. There are no adequate and controlled studies to date using fexofenadine in pregnant women, and fexofenadine hydrochloride alone or in fixed combination with pseudoephedrine hydrochloride should be used during pregnancy only when the potential benefits justify the possible risks to the fetus.
An increased incidence of hypospadias in male infants born to women who received loratadine during pregnancy was reported in one study. However, analysis of data from the National Birth Defects Prevention Study (NBDPS) indicated that use of loratadine during early pregnancy was not associated with an increased risk of second- or third-degree hypospadias. In addition, in 2 small prospective cohort studies that surveyed pregnant women who contacted a teratology information service, use of loratadine during the first trimester of pregnancy was not associated with major congenital anomalies and did not affect the rate of live birth, gestational age at birth, and birth weight.
Despite these findings, it should be noted that interpretation of these results is limited by the statistical limitations of the studies (i.e., small sample size, inadequate power, reliance on patient recall of drug use, exclusion criteria). The 2 prospective cohort studies were powered to detect statistical significance only if a substantial (i.e., approximately threefold) increase in the overall rate of major congenital anomalies was observed; the study that relied on NBDPS data excluded first-degree hypospadias because of the difficulty of detecting this mildest form in routine surveillance, making it difficult to determine the relationship between loratadine and this form of hypospadias. Thus, while these data may be useful, further study is needed to completely rule out the teratogenic risk of loratadine.
Because there are no adequate and controlled studies to date using loratadine in pregnant women, loratadine alone or in fixed combination with pseudoephedrine hydrochloride should be used during pregnancy only when the potential benefits justify the possible risks to the fetus. Reproduction studies in rats and rabbits using loratadine dosages up to 75 and 150 times, respectively, the maximum daily human dosage on a mg/m2 basis have not revealed evidence of harm to the fetus. Reproduction studies in mice, rats, and rabbits using oral cetirizine hydrochloride dosages up to 96, 225, and 135 mg/kg daily, respectively (approximately 40, 180, and 220 times, respectively, the maximum recommended daily oral dosage in adults on a mg/m2 basis), have not revealed evidence of teratogenicity.
Because there are no adequate and controlled studies to date using cetirizine in pregnant women and animal studies are not always predictive of human response, cetirizine hydrochloride should be used during pregnancy only when clearly needed. Cetirizine hydrochloride in combination with pseudoephedrine has been shown to increase the number of fetal skeletal malformations (rib distortions) and variants (unossified sternebrae) in rats when given orally in a fixed-combination ratio at a dosage of 6/154 mg/kg (approximately 5 times the maximum recommended adult dosage on a mg/m2 basis). These effects were not observed at a dosage of 1.6/38
mg/kg (approximately the maximum recommended adult dosage on a mg/m2 basis). Reproduction studies in rabbits using cetirizine hydrochloride and pseudoephedrine hydrochloride in a fixed-combination ratio at a dosage of up to 6/154 mg/kg (approximately 10 times the maximum recommended adult dosage on a mg/m2 basis) have not revealed evidence of harm to the fetus. There are no adequate and controlled studies to date using cetirizine hydrochloride and pseudoephedrine hydrochloride in pregnant women, and the fixed combination should be used during pregnancy only when the potential benefits justify the possible risks to the fetus.
Fluticasone propionate should be used with caution in nursing women, since it is not known whether the drug is distributed into milk in humans. Subcutaneous administration of tritiated fluticasone propionate to lactating rats (10 mcg/kg, less than the maximum recommended daily intranasal dosage in adults on a mcg/m2 basis) resulted in measurable radioactivity in both plasma and milk. Other corticosteroids are distributed into milk and in systemic amounts may cause adverse effects, such as growth suppression, in nursing infants.
Diphenhydramine has been detected in milk. Because of the potential for serious adverse reactions to antihistamines in nursing infants, a decision should be made whether to discontinue nursing or diphenhydramine, taking into account the importance of the drug to the woman. It is not known if fexofenadine hydrochloride is distributed into breast milk; however, pseudoephedrine hydrochloride distributes into breast milk.
Since there are no adequate and controlled studies to date on the use of fexofenadine during lactation in humans and because many drugs are excreted in human milk, the manufacturer states that fexofenadine alone or in fixed combination with pseudoephedrine hydrochloride should be used with caution in nursing women, and a decision should be made whether to discontinue nursing or the drug, taking into account the importance of the drug to the woman. Loratadine and desloratadine distribute readily into breast milk, achieving concentrations that are equivalent to those in plasma (i.e., a milk to plasma AUC ratio of 1.17 and 0.85, respectively). The manufacturer states that about 0.03%
of a single 40-mg dose of loratadine was distributed into breast milk as loratadine and desloratadine over 48 hours. Pseudoephedrine also distributes readily into breast milk. Caution should be exercised when loratadine is administered alone or in fixed combination with pseudoephedrine to a nursing woman, and a decision should be made whether to discontinue nursing or the drug, taking into account the importance of the drug to the woman.
In lactating beagles, about 3% of a cetirizine dose was distributed in milk. In mice, cetirizine caused retarded pup weight gain during lactation when dams were receiving a cetirizine hydrochloride dosage of 96 mg/kg daily (about 40 times the maximum recommended daily dosage in adults on a mg/m2 basis). In rats, cetirizine hydrochloride and pseudoephedrine hydrochloride caused retarded pup weight gain and decreased viability during lactation when administered orally to dams in fixed combination at a dosage of 6/154 mg/kg (approximately 5 times the maximum recommended adult dosage on a mg/m2 basis) but not when administered at a dosage of 1.6/38
mg/kg (approximately the maximum recommended adult dosage on a mg/m2 basis). Cetirizine is distributed into human milk. Pseudoephedrine also distributes into human milk. Therefore, use of cetirizine hydrochloride alone or in combination with pseudoephedrine hydrochloride in nursing women is not recommended.
Diphenhydramine has been detected in milk. Because of the potential for serious adverse reactions to antihistamines in nursing infants, a decision should be made whether to discontinue nursing or diphenhydramine, taking into account the importance of the drug to the woman. It is not known if fexofenadine hydrochloride is distributed into breast milk; however, pseudoephedrine hydrochloride distributes into breast milk.
Since there are no adequate and controlled studies to date on the use of fexofenadine during lactation in humans and because many drugs are excreted in human milk, the manufacturer states that fexofenadine alone or in fixed combination with pseudoephedrine hydrochloride should be used with caution in nursing women, and a decision should be made whether to discontinue nursing or the drug, taking into account the importance of the drug to the woman. Loratadine and desloratadine distribute readily into breast milk, achieving concentrations that are equivalent to those in plasma (i.e., a milk to plasma AUC ratio of 1.17 and 0.85, respectively). The manufacturer states that about 0.03%
of a single 40-mg dose of loratadine was distributed into breast milk as loratadine and desloratadine over 48 hours. Pseudoephedrine also distributes readily into breast milk. Caution should be exercised when loratadine is administered alone or in fixed combination with pseudoephedrine to a nursing woman, and a decision should be made whether to discontinue nursing or the drug, taking into account the importance of the drug to the woman.
In lactating beagles, about 3% of a cetirizine dose was distributed in milk. In mice, cetirizine caused retarded pup weight gain during lactation when dams were receiving a cetirizine hydrochloride dosage of 96 mg/kg daily (about 40 times the maximum recommended daily dosage in adults on a mg/m2 basis). In rats, cetirizine hydrochloride and pseudoephedrine hydrochloride caused retarded pup weight gain and decreased viability during lactation when administered orally to dams in fixed combination at a dosage of 6/154 mg/kg (approximately 5 times the maximum recommended adult dosage on a mg/m2 basis) but not when administered at a dosage of 1.6/38
mg/kg (approximately the maximum recommended adult dosage on a mg/m2 basis). Cetirizine is distributed into human milk. Pseudoephedrine also distributes into human milk. Therefore, use of cetirizine hydrochloride alone or in combination with pseudoephedrine hydrochloride in nursing women is not recommended.
No enhanced Geriatric Use information available for this drug.
The following prioritized warning is available for ALLERGY RELIEF (cetirizine hcl):
No warning message for this drug.
No warning message for this drug.
The following icd codes are available for ALLERGY RELIEF (cetirizine hcl)'s list of indications:
| Allergic conjunctivitis | |
| H10.1 | Acute atopic conjunctivitis |
| H10.10 | Acute atopic conjunctivitis, unspecified eye |
| H10.11 | Acute atopic conjunctivitis, right eye |
| H10.12 | Acute atopic conjunctivitis, left eye |
| H10.13 | Acute atopic conjunctivitis, bilateral |
| H10.44 | Vernal conjunctivitis |
| H10.45 | Other chronic allergic conjunctivitis |
| H16.26 | Vernal keratoconjunctivitis, with limbar and corneal involvement |
| H16.261 | Vernal keratoconjunctivitis, with limbar and corneal involvement, right eye |
| H16.262 | Vernal keratoconjunctivitis, with limbar and corneal involvement, left eye |
| H16.263 | Vernal keratoconjunctivitis, with limbar and corneal involvement, bilateral |
| H16.269 | Vernal keratoconjunctivitis, with limbar and corneal involvement, unspecified eye |
| Allergic reaction | |
| T78.40 | Allergy, unspecified |
| T78.40xA | Allergy, unspecified, initial encounter |
| Allergic rhinitis | |
| J30.1 | Allergic rhinitis due to pollen |
| J30.2 | Other seasonal allergic rhinitis |
| J30.5 | Allergic rhinitis due to food |
| J30.8 | Other allergic rhinitis |
| J30.81 | Allergic rhinitis due to animal (cat) (dog) hair and dander |
| J30.89 | Other allergic rhinitis |
| J30.9 | Allergic rhinitis, unspecified |
| Anaphylaxis | |
| T78.0 | Anaphylactic reaction due to food |
| T78.00 | Anaphylactic reaction due to unspecified food |
| T78.00xA | Anaphylactic reaction due to unspecified food, initial encounter |
| T78.01 | Anaphylactic reaction due to peanuts |
| T78.01xA | Anaphylactic reaction due to peanuts, initial encounter |
| T78.02 | Anaphylactic reaction due to shellfish (crustaceans) |
| T78.02xA | Anaphylactic reaction due to shellfish (crustaceans), initial encounter |
| T78.03 | Anaphylactic reaction due to other fish |
| T78.03xA | Anaphylactic reaction due to other fish, initial encounter |
| T78.04 | Anaphylactic reaction due to fruits and vegetables |
| T78.04xA | Anaphylactic reaction due to fruits and vegetables, initial encounter |
| T78.05 | Anaphylactic reaction due to tree nuts and seeds |
| T78.05xA | Anaphylactic reaction due to tree nuts and seeds, initial encounter |
| T78.06 | Anaphylactic reaction due to food additives |
| T78.06xA | Anaphylactic reaction due to food additives, initial encounter |
| T78.07 | Anaphylactic reaction due to milk and dairy products |
| T78.08 | Anaphylactic reaction due to eggs |
| T78.09 | Anaphylactic reaction due to other food products |
| T78.09xA | Anaphylactic reaction due to other food products, initial encounter |
| T78.2 | Anaphylactic shock, unspecified |
| T78.2xxA | Anaphylactic shock, unspecified, initial encounter |
| T80.5 | Anaphylactic reaction due to serum |
| T80.51 | Anaphylactic reaction due to administration of blood and blood products |
| T80.51xA | Anaphylactic reaction due to administration of blood and blood products, initial encounter |
| T80.52 | Anaphylactic reaction due to vaccination |
| T80.52xA | Anaphylactic reaction due to vaccination, initial encounter |
| T80.59 | Anaphylactic reaction due to other serum |
| T80.59xA | Anaphylactic reaction due to other serum, initial encounter |
| T88.6 | Anaphylactic reaction due to adverse effect of correct drug or medicament properly administered |
| T88.6xxA | Anaphylactic reaction due to adverse effect of correct drug or medicament properly administered, initial encounter |
| Chronic idiopathic urticaria | |
| L50.1 | Idiopathic urticaria |
| Cough | |
| R05 | Cough |
| R05.1 | Acute cough |
| R05.2 | Subacute cough |
| R05.3 | Chronic cough |
| R05.9 | Cough, unspecified |
| Dermatographic urticaria | |
| L50.3 | Dermatographic urticaria |
| Idiopathic parkinsonism | |
| G20 | Parkinson's disease |
| G20.A | Parkinson's disease without dyskinesia |
| G20.A1 | Parkinson's disease without dyskinesia, without mention of fluctuations |
| G20.A2 | Parkinson's disease without dyskinesia, with fluctuations |
| G20.B | Parkinson's disease with dyskinesia |
| G20.B1 | Parkinson's disease with dyskinesia, without mention of fluctuations |
| G20.B2 | Parkinson's disease with dyskinesia, with fluctuations |
| G20.C | Parkinsonism, unspecified |
| Insomnia | |
| F51.0 | Insomnia not due to a substance or known physiological condition |
| F51.01 | Primary insomnia |
| F51.02 | Adjustment insomnia |
| F51.03 | Paradoxical insomnia |
| F51.04 | Psychophysiologic insomnia |
| F51.05 | Insomnia due to other mental disorder |
| F51.09 | Other insomnia not due to a substance or known physiological condition |
| G47.0 | Insomnia |
| G47.00 | Insomnia, unspecified |
| G47.01 | Insomnia due to medical condition |
| G47.09 | Other insomnia |
| Motion sickness | |
| T75.3xxA | Motion sickness, initial encounter |
| Nasal congestion | |
| R09.81 | Nasal congestion |
| Nausea | |
| R11 | Nausea and vomiting |
| R11.0 | Nausea |
| R11.2 | Nausea with vomiting, unspecified |
| Nausea and vomiting | |
| R11 | Nausea and vomiting |
| R11.16 | Cannabis hyperemesis syndrome |
| R11.2 | Nausea with vomiting, unspecified |
| Parkinsonism | |
| G20 | Parkinson's disease |
| G20.A | Parkinson's disease without dyskinesia |
| G20.A1 | Parkinson's disease without dyskinesia, without mention of fluctuations |
| G20.A2 | Parkinson's disease without dyskinesia, with fluctuations |
| G20.B | Parkinson's disease with dyskinesia |
| G20.B1 | Parkinson's disease with dyskinesia, without mention of fluctuations |
| G20.B2 | Parkinson's disease with dyskinesia, with fluctuations |
| G20.C | Parkinsonism, unspecified |
| G21 | Secondary parkinsonism |
| G21.2 | Secondary parkinsonism due to other external agents |
| G21.3 | Postencephalitic parkinsonism |
| G21.4 | Vascular parkinsonism |
| G21.8 | Other secondary parkinsonism |
| G21.9 | Secondary parkinsonism, unspecified |
| Perennial allergic rhinitis | |
| J31.0 | Chronic rhinitis |
| Pruritus of skin | |
| L29.8 | Other pruritus |
| L29.81 | Cholestatic pruritus |
| L29.89 | Other pruritus |
| L29.9 | Pruritus, unspecified |
| Seasonal allergic rhinitis | |
| J30.1 | Allergic rhinitis due to pollen |
| J30.2 | Other seasonal allergic rhinitis |
| Sneezing | |
| R06.7 | Sneezing |
| Urticaria | |
| L50 | Urticaria |
| L50.0 | Allergic urticaria |
| L50.1 | Idiopathic urticaria |
| L50.2 | Urticaria due to cold and heat |
| L50.3 | Dermatographic urticaria |
| L50.4 | Vibratory urticaria |
| L50.5 | Cholinergic urticaria |
| L50.6 | Contact urticaria |
| L50.8 | Other urticaria |
| L50.9 | Urticaria, unspecified |
| L56.3 | Solar urticaria |
| O26.86 | Pruritic urticarial papules and plaques of pregnancy (PUPPp) |
| Vertigo | |
| A88.1 | Epidemic vertigo |
| H81.1 | Benign paroxysmal vertigo |
| H81.10 | Benign paroxysmal vertigo, unspecified ear |
| H81.11 | Benign paroxysmal vertigo, right ear |
| H81.12 | Benign paroxysmal vertigo, left ear |
| H81.13 | Benign paroxysmal vertigo, bilateral |
| H81.2 | Vestibular neuronitis |
| H81.20 | Vestibular neuronitis, unspecified ear |
| H81.21 | Vestibular neuronitis, right ear |
| H81.22 | Vestibular neuronitis, left ear |
| H81.23 | Vestibular neuronitis, bilateral |
| H81.3 | Other peripheral vertigo |
| H81.31 | Aural vertigo |
| H81.311 | Aural vertigo, right ear |
| H81.312 | Aural vertigo, left ear |
| H81.313 | Aural vertigo, bilateral |
| H81.319 | Aural vertigo, unspecified ear |
| H81.39 | Other peripheral vertigo |
| H81.391 | Other peripheral vertigo, right ear |
| H81.392 | Other peripheral vertigo, left ear |
| H81.393 | Other peripheral vertigo, bilateral |
| H81.399 | Other peripheral vertigo, unspecified ear |
| H81.4 | Vertigo of central origin |
| H82 | Vertiginous syndromes in diseases classified elsewhere |
| H82.1 | Vertiginous syndromes in diseases classified elsewhere, right ear |
| H82.2 | Vertiginous syndromes in diseases classified elsewhere, left ear |
| H82.3 | Vertiginous syndromes in diseases classified elsewhere, bilateral |
| H82.9 | Vertiginous syndromes in diseases classified elsewhere, unspecified ear |
| R42 | Dizziness and giddiness |
| T75.23 | Vertigo from infrasound |
| T75.23xA | Vertigo from infrasound, initial encounter |
| Vomiting | |
| K91.0 | Vomiting following gastrointestinal surgery |
| R11 | Nausea and vomiting |
| R11.1 | Vomiting |
| R11.10 | Vomiting, unspecified |
| R11.11 | Vomiting without nausea |
| R11.12 | Projectile vomiting |
| R11.13 | Vomiting of fecal matter |
| R11.14 | Bilious vomiting |
| R11.15 | Cyclical vomiting syndrome unrelated to migraine |
| R11.16 | Cannabis hyperemesis syndrome |
| R11.2 | Nausea with vomiting, unspecified |
Formulary Reference Tool