Please wait while the formulary information is being retrieved.
Drug overview for NEBIVOLOL HCL (nebivolol hcl):
Generic name: NEBIVOLOL HCL (ne-BIV-oh-lol)
Drug class: Beta-Blockers (Systemic)
Therapeutic class: Cardiovascular Therapy Agents
Nebivolol is a beta-adrenergic blocking agent (beta-blocker).
No enhanced Uses information available for this drug.
Generic name: NEBIVOLOL HCL (ne-BIV-oh-lol)
Drug class: Beta-Blockers (Systemic)
Therapeutic class: Cardiovascular Therapy Agents
Nebivolol is a beta-adrenergic blocking agent (beta-blocker).
No enhanced Uses information available for this drug.
DRUG IMAGES
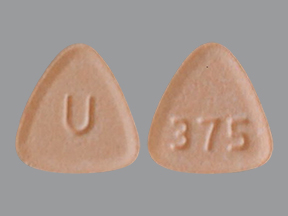 NEBIVOLOL 2.5 MG TABLET
NEBIVOLOL 2.5 MG TABLET NEBIVOLOL 5 MG TABLET
NEBIVOLOL 5 MG TABLET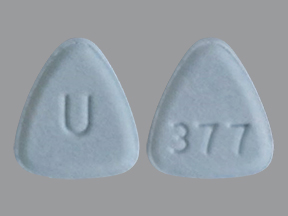 NEBIVOLOL 10 MG TABLET
NEBIVOLOL 10 MG TABLET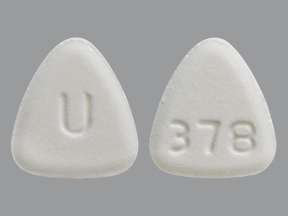 NEBIVOLOL 20 MG TABLET
NEBIVOLOL 20 MG TABLET
The following indications for NEBIVOLOL HCL (nebivolol hcl) have been approved by the FDA:
Indications:
Hypertension
Professional Synonyms:
Elevated blood pressure
Essential hypertension
Hyperpiesia
Hyperpiesis
Hypertensive disorder
Systemic arterial hypertension
Indications:
Hypertension
Professional Synonyms:
Elevated blood pressure
Essential hypertension
Hyperpiesia
Hyperpiesis
Hypertensive disorder
Systemic arterial hypertension
The following dosing information is available for NEBIVOLOL HCL (nebivolol hcl):
Dosage of nebivolol hydrochloride is expressed in terms of nebivolol.
Dosage of nebivolol should be individualized according to the response of the patient. If nebivolol therapy is to be discontinued, dosage of the drug should be reduced gradually over a period of 1-2 weeks when possible.
For the treatment of hypertension in adults, the usual initial dosage of nebivolol is 5 mg once daily as monotherapy or in combination with other antihypertensive agents. In patients whose blood pressure is not controlled adequately with the initial nebivolol dosage, dosage can be increased at 2-week intervals up to 40 mg daily. Some experts state the usual dosage range is 5-40 mg administered once daily.
Administering the drug more frequently (i.e., in divided doses daily) is unlikely to be more beneficial than once-daily administration.
Dosage of nebivolol should be individualized according to the response of the patient. If nebivolol therapy is to be discontinued, dosage of the drug should be reduced gradually over a period of 1-2 weeks when possible.
For the treatment of hypertension in adults, the usual initial dosage of nebivolol is 5 mg once daily as monotherapy or in combination with other antihypertensive agents. In patients whose blood pressure is not controlled adequately with the initial nebivolol dosage, dosage can be increased at 2-week intervals up to 40 mg daily. Some experts state the usual dosage range is 5-40 mg administered once daily.
Administering the drug more frequently (i.e., in divided doses daily) is unlikely to be more beneficial than once-daily administration.
Nebivolol hydrochloride is administered orally once daily without regard to meals.
| DRUG LABEL | DOSING TYPE | DOSING INSTRUCTIONS |
|---|---|---|
| NEBIVOLOL 2.5 MG TABLET | Maintenance | Adults take 2 tablets (5 mg) by oral route once daily |
| NEBIVOLOL 5 MG TABLET | Maintenance | Adults take 1 tablet (5 mg) by oral route once daily |
| NEBIVOLOL 10 MG TABLET | Maintenance | Adults take 1 tablet (10 mg) by oral route once daily |
| NEBIVOLOL 20 MG TABLET | Maintenance | Adults take 1 tablet (20 mg) by oral route once daily |
| DRUG LABEL | DOSING TYPE | DOSING INSTRUCTIONS |
|---|---|---|
| NEBIVOLOL 2.5 MG TABLET | Maintenance | Adults take 2 tablets (5 mg) by oral route once daily |
| NEBIVOLOL 5 MG TABLET | Maintenance | Adults take 1 tablet (5 mg) by oral route once daily |
| NEBIVOLOL 10 MG TABLET | Maintenance | Adults take 1 tablet (10 mg) by oral route once daily |
| NEBIVOLOL 20 MG TABLET | Maintenance | Adults take 1 tablet (20 mg) by oral route once daily |
The following drug interaction information is available for NEBIVOLOL HCL (nebivolol hcl):
There are 1 contraindications.
These drug combinations generally should not be dispensed or administered to the same patient. A manufacturer label warning that indicates the contraindication warrants inclusion of a drug combination in this category, regardless of clinical evidence or lack of clinical evidence to support the contraindication.
| Drug Interaction | Drug Names |
|---|---|
| Selected CYP2D6 Substrates/Mavorixafor SEVERITY LEVEL: 1-Contraindicated Drug Combination: This drug combination is contraindicated and generally should not be dispensed or administered to the same patient. MECHANISM OF ACTION: Mavorixafor is a strong inhibitor of CYP2D6 and is expected to inhibit the metabolism of agents through this pathway.(1) CLINICAL EFFECTS: Concurrent use of mavorixafor may result in elevated levels of and toxicity from agents metabolized by CYP2D6.(1) PREDISPOSING FACTORS: With tricyclic antidepressants, the risk of seizures may be increased in patients with a history of head trauma or prior seizure; CNS tumor; severe hepatic cirrhosis; excessive use of alcohol or sedatives; addiction to opiates, cocaine, or stimulants; use of over-the-counter stimulants and anorectics; diabetics treated with oral hypoglycemics or insulin; or with concomitant medications known to lower seizure threshold (antipsychotics, theophylline, systemic steroids). With anticholinergic agents, the risk of anticholinergic toxicities including cognitive decline, delirium, falls and fractures is increased in geriatric patients using more than one medicine with anticholinergic properties.(2) PATIENT MANAGEMENT: The US manufacturer of mavorixafor states concurrent use with CYP2D6 substrate that are highly dependent on CYP2D6 metabolism is contraindicated.(1) The US manufacturer of doxepin states if concurrent use of doxepin and strong CYP2D6 inhibitors such as mavorixafor is warranted, monitor doxepin plasma concentrations and reduce the doxepin dose based on doxepin plasma concentrations.(5) DISCUSSION: Mavorixafor (400 mg) increased dextromethorphan (CYP2D6 substrate) maximum concentration (Cmax) and area-under-curve (AUC) by 6-fold and 9-fold, respectively.(1) Selected CYP2D6 substrates linked to this monograph include: aripiprazole, atomoxetine, brexpiprazole, desipramine, deutetrabenazine, dextromethorphan, doxepin, encainide, fenfluramine, metoclopramide, methoxyphenamine, metoprolol, mexiletine, nebivolol, paroxetine, perphenazine, risperidone, tetrabenazine, trimipramine, venlafaxine, and yohimbine. |
XOLREMDI |
There are 8 severe interactions.
These drug interactions can produce serious consequences in most patients. Actions required for severe interactions include, but are not limited to, discontinuing one or both agents, adjusting dosage, altering administration scheduling, and providing additional patient monitoring. Review the full interaction monograph for more information.
| Drug Interaction | Drug Names |
|---|---|
| Fingolimod/Beta-Blockers; AV Node Blockers SEVERITY LEVEL: 2-Severe Interaction: Action is required to reduce the risk of severe adverse interaction. MECHANISM OF ACTION: Initiation of fingolimod has a negative chronotropic effect leading to a mean decrease in heart rate of 13 beats per minute (bpm) after the first dose. The first dose has also been associated with heart block. Beta-blockers or agents which slow AV node conduction further increase the risk for symptomatic bradycardia or heart block. CLINICAL EFFECTS: The heart rate lowering effect of fingolimod is biphasic with an initial decrease usually within 6 hours, followed by a second decrease 12 to 24 hours after the first dose. Symptomatic bradycardia and heart block have been observed. Bradycardia may be associated with an increase in the QTc interval, increasing the risk for torsade de pointes. The cause of death in a patient who died within 24 hour after taking the first dose of fingolimod was not conclusive; however a link to fingolimod or a drug interaction with fingolimod could not be ruled out. Beta-Blockers linked to this monograph are: atenolol, betaxolol, bisoprolol, carvedilol, esmolol, landiolol, labetalol, metoprolol, nadolol, nebivolol, propranolol and timolol. AV Node Blocking agents are:digoxin, diltiazem, flecainide, ivabradine, propafenone and verapamil. PREDISPOSING FACTORS: Pre-existing cardiovascular disease (e.g. heart failure, ischemic heart disease, history of myocardial infarction, stroke, history of torsades de pointes, or heart block), severe untreated sleep apnea, a prolonged QTc interval prior to fingolimod initiation, or factors associated with QTc prolongation (e.g. hypokalemia, hypomagnesemia, bradycardia, female gender, or advanced age) may increase risk for cardiovascular toxicity due to fingolimod. Concurrent use of more than one drug known to cause QT prolongation or higher systemic concentrations of either QT prolonging drug are additional risk factors for torsades de pointes. Factors which may increase systemic drug concentrations include rapid infusion of an intravenous dose or impaired metabolism or elimination of the drug (e.g. coadministration with an agent which inhibits its metabolism or elimination, genetic impairment in drug metabolism or elimination, and/or renal/hepatic dysfunction).(5) PATIENT MANAGEMENT: Fingolimod is contraindicated in patients with Class III/IV heart failure or in patients who have experienced myocardial infarction, unstable angina, stroke, transient ischemic attack (TIA) or decompensated heart failure within the past six months.(1) Patients with pre-existing cardiovascular or cerebrovascular disease (e.g. heart failure, ischemic heart disease, history of myocardial infarction, stroke, or heart block), severe untreated sleep apnea, or a prolonged QTc interval prior to fingolimod initiation should receive cardiologist consultation to evaluate the risks of fingolimod therapy. Patients receiving agents linked to this monograph should have their physician evaluate the possibility of a switch to agents which do not slow heart rate or cardiac conduction. If fingolimod is initiated, the patient should stay overnight in a medical facility with continuous ECG monitoring after the first dose. Correct hypokalemia or hypomagnesemia prior to starting fingolimod. US monitoring recommendations in addition to continuous ECG with overnight monitoring: Check blood pressure hourly. If heart rate (HR) is < 45 beats per minute (BPM) or if the ECG shows new onset of second degree or higher AV block at the end of the monitoring period, then monitoring should continue until the finding has resolved. If patient requires treatment for symptomatic bradycardia, the first dose monitoring strategy should be repeated for the second dose of fingolimod. If, within the first two weeks of treatment one or more fingolimod doses is missed, then first dose procedures are recommended upon resumption. If during weeks 3 and 4 of fingolimod treatment dose is interrupted more than 7 days, then first dose procedures are recommended upon resumption. United Kingdom recommendations(3): Obtain a 12-lead ECG prior to initiating fingolimod therapy. Consult a cardiologist for pretreatment risk-benefit assessment if patient has a resting heart rate less than 55 bpm, history of syncope, second degree or greater AV block, sick-sinus syndrome, concurrent therapy with beta-blockers, Class Ia, or Class III antiarrhythmics, heart failure or other significant cardiovascular disease. Perform continuous ECG monitoring, measure blood pressure and heart rate every hour, and perform a 12-lead ECG 6 hours after the first dose. Monitoring should be extended beyond 6 hours if symptomatic bradycardia or new onset of second degree AV block, Mobitz Type II or third degree AV block has occurred at any time during the monitoring period. If heart rate 6 hours after the first dose is less than 40 bpm, has decreased more than 20 bpm compared with baseline, or if a new onset second degree AV block, Mobitz Type I (Wenckebach) persists, then monitoring should also be continued. If fingolimod treatment is discontinued for more than two weeks, the effects on heart rate and conduction could recur. Thus, first dose monitoring precautions should be followed upon reintroduction of fingolimod. DISCUSSION: After the first dose of fingolimod, heart rate decrease may begin within an hour. Decline is usually maximal at approximately 6 hours followed by a second decrease 12 to 24 hours after the first dose. The second dose may further decrease heart rate, but the magnitude of change is smaller than the first dose. With continued, chronic dosing, heart rate gradually returns to baseline in about one month.(1,2) Diurnal variation in heart rate and response to exercise are not affected by fingolimod treatment.(2) In a manufacturer sponsored study, fingolimod and atenolol 50 mg daily lowered heart rate 15% more than fingolimod alone. However, additional heart rate lowering was not seen with the combination of extended release diltiazem and fingolimod compared with fingolimod alone.(1) |
FINGOLIMOD, GILENYA, TASCENSO ODT |
| Allergen Immunotherapy/Beta-Blockers SEVERITY LEVEL: 2-Severe Interaction: Action is required to reduce the risk of severe adverse interaction. MECHANISM OF ACTION: Beta-blockers may mask early signs and symptoms of anaphylaxis, make the treatment of anaphylaxis more difficult, and increase the severity of the reaction. CLINICAL EFFECTS: Beta-blockers may reduce a patient's ability to survive a systemic allergic reaction to allergen immunotherapy. Signs and symptoms of anaphylaxis may be masked. PREDISPOSING FACTORS: Concurrent use of epinephrine with beta-blockers may result in hypertension with reflex bradycardia. Epinephrine resistance in patients with anaphylaxis has been reported. PATIENT MANAGEMENT: Avoid concomitant administration of immunotherapy and beta-blockers if possible. If patients cannot safely discontinue beta-blockers but have a history of moderate to severe sting-induced anaphylaxis, venom immunotherapy is indicated because the risk of anaphylaxis related to a venom sting is greater than the risk of an immunotherapy-related systemic reaction. In patients taking beta-blockers for whom an acceptable alternative is not available, withholding allergen immunotherapy may be the best option. If both drugs are administered, monitor closely for signs and symptoms of anaphylaxis. Use caution when treating anaphylaxis with epinephrine since response may be poor. Epinephrine administration may worsen anaphylaxis because beta-blockers block the beta effects of epinephrine, which results in predomination of alpha effects. The plasma clearance of epinephrine is decreased. Glucagon may help in the treatment of refractory anaphylaxis in patients receiving beta-blockers. DISCUSSION: In a case report, a patient taking propranolol was administered pollen extract immunotherapy and immediately developed anaphylaxis. Treatment with epinephrine did not improve symptoms and patient was subsequently intubated.(2) In another case report, a patient taking propranolol was given pollen immunotherapy and developed anaphylaxis. Difficulty in maintaining an adequate blood pressure and pulse continued for several hours despite epinephrine and other supportive measures.(3) There are other case reports of patients taking propranolol with venom immunotherapy that were refractory to treatment.(6-7) |
9 TREE MIX EXTRACT, ACACIA, ALDER, ALFALFA EXTRACT, ALTERNARIA ALTERNATA, AMERICAN BEECH, AMERICAN COCKROACH EXTRACT, AMERICAN ELM, AMERICAN SYCAMORE, ARIZONA CYPRESS, ASPERGILLUS FUMIGATUS, AUREOBASIDIUM PULLULANS, BAHIA, BALD CYPRESS, BAYBERRY, BLACK WALNUT POLLEN, BOTRYTIS CINEREA, BOX ELDER, BROME, CALIFORNIA PEPPER TREE, CANDIDA ALBICANS, CARELESSWEED, CATTLE EPITHELIUM, CEDAR ELM, CLADOSPORIUM CLADOSPORIOIDES, COCKLEBUR, CORN POLLEN, CORN SMUT, D.FARINAE MITE EXTRACT, D.PTERONYSSINUS MITE EXTRACT, DOG EPITHELIUM EXTRACT, DOG FENNEL, EASTERN COTTONWOOD, ENGLISH PLANTAIN, EPICOCCUM NIGRUM, FIRE ANT, GERMAN COCKROACH, GOLDENROD, GRASTEK, GUINEA PIG EPITHELIUM EXTRACT, HACKBERRY, HONEY BEE VENOM PROTEIN, HORSE EPITHELIUM, JOHNSON GRASS, KOCHIA, LAMB'S QUARTERS, MELALEUCA, MESQUITE, MIXED COCKROACH, MIXED FEATHERS, MIXED RAGWEED EXTRACT, MIXED VESPID VENOM PROTEIN, MOSQUITO, MOUNTAIN CEDAR, MOUSE EPITHELIUM, MUCOR PLUMBEUS, MUGWORT, ODACTRA, OLIVE TREE, ORALAIR, PALFORZIA, PECAN POLLEN, PENICILLIUM NOTATUM, PRIVET, QUACK GRASS, QUEEN PALM, RABBIT EPITHELIUM, RAGWITEK, RED BIRCH, RED CEDAR, RED MAPLE, RED MULBERRY, RED OAK, ROUGH MARSH ELDER, ROUGH PIGWEED, RUSSIAN THISTLE, SACCHAROMYCES CEREVISIAE, SAGEBRUSH, SAROCLADIUM STRICTUM, SHAGBARK HICKORY, SHEEP SORREL, SHEEP SORREL-YELLOW DOCK, SHORT RAGWEED, SPINY PIGWEED, STANDARD BERMUDA GRASS POLLEN, STANDARD MIXED GRASS POLLEN, STANDARD MIXED MITE EXTRACT, STANDARD RYE GRASS POLLEN, STANDARD SWEET VERNAL GRASS, STANDARDIZED CAT HAIR, STANDARDIZED JUNE GRASS POLLEN, STANDARDIZED MEADOW FESCUE, STANDARDIZED ORCHARD GRASS, STANDARDIZED RED TOP GRASS, STANDARDIZED TIMOTHY GRASS, SWEETGUM, TALL RAGWEED, TRICHOPHYTON MENTAGROPHYTES, VIRGINIA LIVE OAK, WASP VENOM PROTEIN, WEED MIX NO.7B EXTRACT, WESTERN JUNIPER, WESTERN RAGWEED, WHITE ASH, WHITE BIRCH, WHITE MULBERRY, WHITE OAK EXTRACT, WHITE PINE, WHITE-FACED HORNET VENOM, YELLOW DOCK, YELLOW HORNET VENOM PROTEIN, YELLOW JACKET VENOM PROTEIN |
| Siponimod/Beta-Blockers SEVERITY LEVEL: 2-Severe Interaction: Action is required to reduce the risk of severe adverse interaction. MECHANISM OF ACTION: Initiation of siponimod has caused transient decreases in heart rate and atrioventricular conduction delays after the first dose. Decreases in heart rate start within the first hour and maximal decrease in heart rate was seen at approximately 3-4 hours. The first dose has also been associated with heart block. Beta-blockers further increase the risk for symptomatic bradycardia or heart block.(1) CLINICAL EFFECTS: The heart rate lowering effect of siponimod is transient and is usually seen with the first dose. Bradycardia may be associated with an increase in the QTc interval, increasing the risk for torsade de pointes.(1) PREDISPOSING FACTORS: Pre-existing cardiovascular disease (e.g. heart failure, ischemic heart disease, history of myocardial infarction, stroke, history of torsades de pointes, or heart block), severe untreated sleep apnea, a prolonged QTc interval prior to siponimod initiation, or factors associated with QTc prolongation (e.g. hypokalemia, hypomagnesemia, bradycardia, female gender, or advanced age) may increase risk for cardiovascular toxicity due to siponimod. Concurrent use of more than one drug known to cause QT prolongation or higher systemic concentrations of either QT prolonging drug are additional risk factors for torsades de pointes. Factors which may increase systemic drug concentrations include rapid infusion of an intravenous dose or impaired metabolism or elimination of the drug (e.g. coadministration with an agent which inhibits its metabolism or elimination, genetic impairment in drug metabolism or elimination, and/or renal/hepatic dysfunction).(2) PATIENT MANAGEMENT: The prescribing information states temporary interruption in beta-blocker therapy may be needed before initiation of siponimod. Beta-blocker therapy can be initiated in patients receiving stable doses of siponimod.(1) Treatment initiation recommendations include: - Obtain an ECG in all patients to determine whether preexisting conduction abnormalities are present. - In all patients, a dose titration is recommended for initiation of siponimod treatment to help reduce cardiac effects. - In patients with sinus bradycardia (HR less than 55 bpm), first- or second-degree [Mobitz type I] AV block, or a history of myocardial infarction or heart failure with onset > 6 months prior to initiation, ECG testing and first dose monitoring is recommended. - Since significant bradycardia may be poorly tolerated in patients with history of cardiac arrest, cerebrovascular disease, uncontrolled hypertension, or severe untreated sleep apnea, siponimod is not recommended in these patients. If treatment is considered, advice from a cardiologist should be sought prior to initiation of treatment in order to determine the most appropriate monitoring strategy. - Use of siponimod in patients with a history of recurrent syncope or symptomatic bradycardia should be based on an overall benefit-risk assessment. If treatment is considered, advice from a cardiologist should be sought prior to initiation of treatment in order to determine the most appropriate monitoring. - For patients receiving a stable dose of a beta-blocker, the resting heart rate should be considered before introducing siponimod treatment. If the resting heart rate is greater than 50 bpm under chronic beta-blocker treatment, siponimod can be introduced. If resting heart rate is less than or equal to 50 bpm, beta-blocker treatment should be interrupted until the baseline heart-rate is greater than 50 bpm. Treatment with siponimod can then be initiated and treatment with a beta-blocker can be reinitiated after siponimod has been up-titrated to the target maintenance dosage. - If a titration dose is missed or if 4 or more consecutive daily doses are missed during maintenance treatment, reinitiate Day 1 of the dose titration and follow titration monitoring recommendations.(1) DISCUSSION: After the first titration dose of siponimod, the heart rate decrease starts within an hour, and the Day 1 decline is maximal at approximately 3-4 hours. With continued up-titration, further heart rate decreases are seen on subsequent days, with maximal decrease from Day 1-baseline reached on Day 5-6. The highest daily post-dose decrease in absolute hourly mean heart rate is observed on Day 1, with the pulse declining on average 5-6 bpm. Post-dose declines on the following days are less pronounced. With continued dosing, heart rate starts increasing after Day 6 and reaches placebo levels within 10 days after treatment initiation. In Study 1, bradycardia occurred in 4.4% of siponimod-treated patients compared to 2.9% of patients receiving placebo. Patients who experienced bradycardia were generally asymptomatic. Few patients experienced symptoms, including dizziness or fatigue, and these symptoms resolved within 24 hours without intervention.(1) Beta-Blockers linked to this monograph are: atenolol, betaxolol, bisoprolol, carvedilol, esmolol, landiolol, labetalol, metoprolol, nadolol, nebivolol, propranolol and timolol. |
MAYZENT |
| Selected CYP2D6 Substrates/Panobinostat SEVERITY LEVEL: 2-Severe Interaction: Action is required to reduce the risk of severe adverse interaction. MECHANISM OF ACTION: Panobinostat is a moderate inhibitor of CYP2D6 and is expected to inhibit the metabolism of agents through this pathway.(1) CLINICAL EFFECTS: Concurrent use of panobinostat may result in elevated levels of and toxicity from agents metabolized by CYP2D6.(1) PREDISPOSING FACTORS: With tricyclic antidepressants, the risk of seizures may be increased in patients with a history of head trauma or prior seizure; CNS tumor; severe hepatic cirrhosis; excessive use of alcohol or sedatives; addiction to opiates, cocaine, or stimulants; use of over-the-counter stimulants and anorectics; diabetics treated with oral hypoglycemics or insulin; or with concomitant medications known to lower seizure threshold (antipsychotics, theophylline, systemic steroids). With anticholinergic agents, the risk of anticholinergic toxicities including cognitive decline, delirium, falls and fractures is increased in geriatric patients using more than one medicine with anticholinergic properties.(4) PATIENT MANAGEMENT: Avoid the concurrent use of panobinostat with agents that are sensitive CYP2D6 or CYP2D6 substrates with a narrow therapeutic index. If concurrent use is warranted, monitor patients for toxicity.(1) DISCUSSION: In a study in 14 subjects with advanced cancer, panobinostat (20 mg daily on Days 3, 5, and 8) increased the maximum concentration (Cmax) and area-under-curve (AUC) of a single dose of dextromethorphan (60 mg) by 20-200% and 20-130%, respectively. Dextromethorphan exposures were extremely variable.(1) Selected CYP2D6 substrates linked to this monograph include: atomoxetine, desipramine, deutetrabenazine, dextromethorphan, doxepin, encainide, methoxyphenamine, metoprolol, nebivolol, paroxetine, perphenazine, risperidone, tetrabenazine, trimipramine, venlafaxine, and yohimbine. |
FARYDAK |
| Crizotinib/Agents That Cause Bradycardia SEVERITY LEVEL: 2-Severe Interaction: Action is required to reduce the risk of severe adverse interaction. MECHANISM OF ACTION: Crizotinib may cause symptomatic bradycardia. Additional agents that cause bradycardia further increase the risk for symptomatic bradycardia.(1) CLINICAL EFFECTS: Bradycardia may be associated with an increase in the QTc interval, increasing the risk for torsade de pointes.(1) PREDISPOSING FACTORS: None determined. PATIENT MANAGEMENT: The manufacturer of crizotinib recommends avoiding concurrent use of crizotinib and other agents known to cause bradycardia to the extent possible. If combination therapy is required, monitor heart rate and blood pressure regularly. If bradycardia occurs, withhold crizotinib until heart rate recovers to 60 bpm or above, or patient is asymptomatic. Re-evaluate the use of the concomitant medication. If the concomitant medication is discontinued or its dose is reduced, resume crizotinib at the previous dose upon patient's recovery. If the concomitant medication is not discontinued or dose adjusted, resume crizotinib at a reduced dose upon patient's recovery. If life-threatening bradycardia occurs, discontinue or reduce the dose of the concomitant medication. Upon the patient's recovery, lower the dose of crizotinib to 250 mg daily. Monitor blood pressure and heart rate frequently.(1) DISCUSSION: Across clinical trials, bradycardia occurred in 13 % of patients on crizotinib, and grade 3 syncope occurred in 2.4 % of patients on crizotinib compared with 0.6 % on chemotherapy.(1) Agents that may cause bradycardia and linked to this monograph include: beta-blockers, non-dihydropyridine calcium channel blockers, clonidine, digoxin, and moxonidine.(1) |
XALKORI |
| Selected CYP1A2 or CYP2D6 Substrates/Givosiran SEVERITY LEVEL: 2-Severe Interaction: Action is required to reduce the risk of severe adverse interaction. MECHANISM OF ACTION: Givosiran interferes with the first and rate-limiting step in hepatic heme biosynthesis, which may lower hepatic heme levels and decrease production and/or activity of cytochrome P450 enzymes.(1,2) CLINICAL EFFECTS: Concurrent use of givosiran may result in elevated levels of and toxicity from agents metabolized by CYP1A2 or CYP2D6.(1) PREDISPOSING FACTORS: With tricyclic antidepressants, the risk of seizures may be increased in patients with a history of head trauma or prior seizure; CNS tumor; severe hepatic cirrhosis; excessive use of alcohol or sedatives; addiction to opiates, cocaine, or stimulants; use of over-the-counter stimulants and anorectics; diabetics treated with oral hypoglycemics or insulin; or with concomitant medications known to lower seizure threshold (antipsychotics, theophylline, systemic steroids). With anticholinergic agents, the risk of anticholinergic toxicities including cognitive decline, delirium, falls and fractures is increased in geriatric patients using more than one medicine with anticholinergic properties.(3) PATIENT MANAGEMENT: Avoid the concurrent use of givosiran with agents that are sensitive substrates of CYP1A2 or CYP2D6, or CYP1A2 or CYP2D6 substrates with a narrow therapeutic index. If concurrent use is unavoidable, decrease the dose of the CYP1A2 or CYP2D6 substrate and monitor patients for toxicity. DISCUSSION: A study of 9 patients with acute intermittent porphyria found that givosiran decreased the maximum concentration (Cmax) and area-under-curve (AUC) of caffeine (a CYP1A2 substrate) by 1.3- and 3.1-fold, respectively, compared to caffeine alone. Givosiran also decreased the Cmax and AUC of dextromethorphan (a CYP2D6 substrate) by 2- and 2.4-fold, respectively, compared to dextromethorphan alone.(1,2) Selected CYP2D6 substrates linked to this monograph include: atomoxetine, desipramine, deutetrabenazine, dextromethorphan, doxepin, encainide, methoxyphenamine, metoprolol, nebivolol, nefazodone, paroxetine, perphenazine, risperidone, tetrabenazine, trimipramine, and venlafaxine. Selected CYP1A2 substrates linked to this monograph include: agomelatine, aminophylline, rasagiline, tacrine, theophylline, tizanidine, and yohimbine. |
GIVLAARI |
| Ponesimod/Beta-Blockers SEVERITY LEVEL: 2-Severe Interaction: Action is required to reduce the risk of severe adverse interaction. MECHANISM OF ACTION: Initiation of ponesimod has caused transient decreases in heart rate and atrioventricular conduction delays after the first dose. Decreases in heart rate start within the first hour and maximal decrease in heart rate was seen at approximately 2-4 hours. The first dose has also been associated with heart block. Beta-blockers further increase the risk for symptomatic bradycardia or heart block.(1) CLINICAL EFFECTS: The heart rate lowering effect of ponesimod is transient and is usually seen with the first dose. Bradycardia may be associated with an increase in the QTc interval, increasing the risk for torsade de pointes.(1) PREDISPOSING FACTORS: Pre-existing cardiovascular disease (e.g. heart failure, ischemic heart disease, history of myocardial infarction, stroke, history of torsades de pointes, or heart block), severe untreated sleep apnea, a prolonged QTc interval prior to siponimod initiation, or factors associated with QTc prolongation (e.g. hypokalemia, hypomagnesemia, bradycardia, female gender, or advanced age) may increase risk for cardiovascular toxicity due to siponimod. Concurrent use of more than one drug known to cause QT prolongation or higher systemic concentrations of either QT prolonging drug are additional risk factors for torsades de pointes. Factors which may increase systemic drug concentrations include rapid infusion of an intravenous dose or impaired metabolism or elimination of the drug (e.g. coadministration with an agent which inhibits its metabolism or elimination, genetic impairment in drug metabolism or elimination, and/or renal/hepatic dysfunction).(2) PATIENT MANAGEMENT: The prescribing information states temporary interruption in beta-blocker therapy may be needed before initiation of ponesimod. Beta-blocker therapy can be initiated in patients receiving stable doses of ponesimod.(1) Treatment initiation recommendations include: - Obtain an ECG in all patients to determine whether preexisting conduction abnormalities are present. - In all patients, a dose titration is recommended for initiation of ponesimod treatment to help reduce cardiac effects. - In patients with sinus bradycardia (HR less than 55 bpm), first- or second-degree [Mobitz type I] AV block, or a history of myocardial infarction or heart failure with onset > 6 months prior to initiation, ECG testing and first dose monitoring is recommended. - Since significant bradycardia may be poorly tolerated in patients with history of cardiac arrest, cerebrovascular disease, uncontrolled hypertension, or severe untreated sleep apnea, ponesimod is not recommended in these patients. If treatment is considered, advice from a cardiologist should be sought prior to initiation of treatment in order to determine the most appropriate monitoring strategy. - Use of ponesimod in patients with a history of recurrent syncope or symptomatic bradycardia should be based on an overall benefit-risk assessment. If treatment is considered, advice from a cardiologist should be sought prior to initiation of treatment in order to determine the most appropriate monitoring. - For patients receiving a stable dose of a beta-blocker, the resting heart rate should be considered before introducing ponesimod treatment. If the resting heart rate is greater than 55 bpm under chronic beta-blocker treatment, ponesimod can be introduced. If resting heart rate is less than or equal to 55 bpm, beta-blocker treatment should be interrupted until the baseline heart-rate is greater than 55 bpm. Treatment with ponesimod can then be initiated and treatment with a beta-blocker can be reinitiated after ponesimod has been up-titrated to the target maintenance dosage. - If a titration dose is missed or if 4 or more consecutive daily doses are missed during maintenance treatment, reinitiate Day 1 of the dose titration and follow titration monitoring recommendations.(1) DISCUSSION: After the first titration dose of ponesimod the heart rate decrease starts within an hour, and the Day 1 decline is maximal at approximately 2-4 hours. With continued up-titration, further heart rate decreases are seen on subsequent days, with maximal decrease from Day 1-baseline reached on Day 4-5. The highest daily post-dose decrease in absolute hourly mean heart rate is observed on Day 1, with the pulse declining on average 6 bpm. Post-dose declines on the following days are less pronounced. With continued dosing, heart rate starts increasing after Day 6 and reaches placebo levels within 10 days after treatment initiation. In a study, bradycardia occurred in 5.8% of ponesimod-treated patients compared to 1.6% of patients receiving placebo. Patients who experienced bradycardia were generally asymptomatic. Few patients experienced symptoms, including dizziness or fatigue, and these symptoms resolved within 24 hours without intervention.(1) Beta-Blockers linked to this monograph are: atenolol, betaxolol, bisoprolol, carvedilol, esmolol, landiolol, labetalol, metoprolol, nadolol, nebivolol, propranolol and timolol. |
PONVORY |
| Etrasimod/Beta-Blockers SEVERITY LEVEL: 2-Severe Interaction: Action is required to reduce the risk of severe adverse interaction. MECHANISM OF ACTION: Initiation of etrasimod has caused transient decreases in heart rate and atrioventricular conduction delays after the first dose. The first dose has also been associated with heart block. Beta-blockers further increase the risk for symptomatic bradycardia or heart block.(1) CLINICAL EFFECTS: The heart rate lowering effect of etrasimod is transient and is usually seen with the first dose. Bradycardia may be associated with an increase in the QTc interval, increasing the risk for torsade de pointes.(1) PREDISPOSING FACTORS: Pre-existing cardiovascular disease (e.g. heart failure, ischemic heart disease, history of myocardial infarction, stroke, history of torsades de pointes, or heart block), severe untreated sleep apnea, a prolonged QTc interval prior to etrasimod initiation, or factors associated with QTc prolongation (e.g. hypokalemia, hypomagnesemia, bradycardia, female gender, or advanced age) may increase risk for cardiovascular toxicity due to etrasimod. Concurrent use of more than one drug known to cause QT prolongation or higher systemic concentrations of either QT prolonging drug are additional risk factors for torsades de pointes. Factors which may increase systemic drug concentrations include rapid infusion of an intravenous dose or impaired metabolism or elimination of the drug (e.g. coadministration with an agent which inhibits its metabolism or elimination, genetic impairment in drug metabolism or elimination, and/or renal/hepatic dysfunction).(2) PATIENT MANAGEMENT: The prescribing information states etrasimod therapy can be initiated in patients receiving stable doses of beta blocker therapy. Cardiology consultation is recommended before initiating a beta blocker in a patient receiving stable etrasimod treatment.(1) DISCUSSION: Initiation of etrasimod may result in a transient decrease in heart rate and AV conduction delays. In two studies, after the first dose of etrasimod 2 mg, ulcerative colitis patients saw a mean decrease from baseline in heart rate of 7.2 bpm at hour 3 in UC-1 an hour 2 in UC-2.(1) In UC-1, bradycardia was reported on Day 1 in 1% of etrasimod patients, 0.3% on Day 2 compared to no patients receiving placebo.In UC-2 and UC-3, bradycardia was reported on Day 1 in 2.9% of etrasimod patients, 0.3% on Day 2 compared to no patients receiving placebo. Patients experiencing bradycardia were generally asymptomatic. The few patients with symptomatic bradycardia reported dizziness that resolved without intervention.(1) Beta-Blockers linked to this monograph are: atenolol, betaxolol, bisoprolol, carvedilol, esmolol, labetalol, landiolol, metoprolol, nadolol, nebivolol, propranolol and timolol. |
VELSIPITY |
There are 18 moderate interactions.
The clinician should assess the patient’s characteristics and take action as needed. Actions required for moderate interactions include, but are not limited to, discontinuing one or both agents, adjusting dosage, altering administration.
| Drug Interaction | Drug Names |
|---|---|
| Lidocaine/Beta-Blockers SEVERITY LEVEL: 3-Moderate Interaction: Assess the risk to the patient and take action as needed. MECHANISM OF ACTION: Decreased cardiac output and hepatic blood flow due to beta-blockers may result in reduced elimination of lidocaine. Inhibition of hepatic microsomal enzymes may also contribute to decreased lidocaine clearance. CLINICAL EFFECTS: Lidocaine toxicity is more likely to occur when these drugs are used in combination. PREDISPOSING FACTORS: None determined. PATIENT MANAGEMENT: The lidocaine dose may need to be adjusted when a beta-blocker is added or discontinued. Clinical signs of lidocaine toxicity and lidocaine plasma levels should be monitored. DISCUSSION: The effect seems to be more pronounced in the lipid soluble beta-blockers (eg. propranolol, metoprolol). |
LIDOCAINE, LIDOCAINE HCL, LIDOCAINE HCL IN 5% DEXTROSE |
| Selected Beta-blockers/Selected Calcium Channel Blockers SEVERITY LEVEL: 3-Moderate Interaction: Assess the risk to the patient and take action as needed. MECHANISM OF ACTION: Synergistic pharmacologic activity. CLINICAL EFFECTS: May see an increase in the therapeutic and toxic effects of both drugs. Concurrent use in patients with low heart rates may unmask sick sinus syndrome. PREDISPOSING FACTORS: Preexisting left ventricular dysfunction and high doses of the beta-blocking agent may predispose patients to adverse responses to this drug combination. Other possible factors include parenteral administration and concurrent administration of other cardio-depressant drugs such as antiarrhythmics. PATIENT MANAGEMENT: Monitor the patient for signs of increased cardio-depressant effects and hypotension. Adjust the dose accordingly. DISCUSSION: Coadministration of these classes of drugs may be effective in the treatment of angina pectoris and hypertension. Patients should be screened in order to determine who should receive this combination of agents. The concurrent use of mibefradil and beta-blockers in patients with low heart rates may unmask underlying sick sinus syndrome. One or more of the drug pairs linked to this monograph have been included in a list of interactions that could be considered for classification as "non-interruptive" in EHR systems. This DDI subset was vetted by an expert panel commissioned by the U.S. Office of the National Coordinator (ONC) for Health Information Technology. |
CARDIZEM, CARDIZEM CD, CARDIZEM LA, CARTIA XT, DILT-XR, DILTIAZEM 12HR ER, DILTIAZEM 24HR ER, DILTIAZEM 24HR ER (CD), DILTIAZEM 24HR ER (LA), DILTIAZEM 24HR ER (XR), DILTIAZEM HCL, DILTIAZEM HCL-0.7% NACL, DILTIAZEM HCL-0.9% NACL, DILTIAZEM HCL-NACL, DILTIAZEM-D5W, MATZIM LA, NIFEDIPINE, NIFEDIPINE ER, NIFEDIPINE MICRONIZED, PROCARDIA XL, TIADYLT ER, TIAZAC, TRANDOLAPRIL-VERAPAMIL ER, VERAPAMIL ER, VERAPAMIL ER PM, VERAPAMIL HCL, VERAPAMIL SR |
| NSAIDs; Aspirin (Non-Cardioprotective)/Beta-Blockers SEVERITY LEVEL: 3-Moderate Interaction: Assess the risk to the patient and take action as needed. MECHANISM OF ACTION: Unknown; however, possibly related to inhibition of prostaglandin by NSAIDs. CLINICAL EFFECTS: The antihypertensive action of beta-blockers may be decreased. PREDISPOSING FACTORS: None determined. PATIENT MANAGEMENT: Monitor patient's blood pressure and adjust the dose of the beta-blocker as needed. DISCUSSION: Concurrent administration of beta-blockers and NSAIDs has been associated with a clinically significant loss in antihypertensive response. The magnitude of the effect of NSAIDs on control of blood pressure by beta-blockers needs to be determined for each anti-inflammatory agent. One or more of the drug pairs linked to this monograph have been included in a list of interactions that could be considered for classification as "non-interruptive" in EHR systems. This DDI subset was vetted by an expert panel commissioned by the U.S. Office of the National Coordinator (ONC) for Health Information Technology. |
ACETYL SALICYLIC ACID, ANAPROX DS, ARTHROTEC 50, ARTHROTEC 75, ASA-BUTALB-CAFFEINE-CODEINE, ASCOMP WITH CODEINE, ASPIRIN, BISMUTH SUBSALICYLATE, BROMFENAC SODIUM, BUPIVACAINE-KETOROLAC-KETAMINE, BUTALBITAL-ASPIRIN-CAFFEINE, CALDOLOR, CAMBIA, CARISOPRODOL-ASPIRIN, CARISOPRODOL-ASPIRIN-CODEINE, CELEBREX, CELECOXIB, CHOLINE MAGNESIUM TRISALICYLAT, COMBOGESIC, COMBOGESIC IV, CONSENSI, COXANTO, DICLOFENAC, DICLOFENAC POTASSIUM, DICLOFENAC SODIUM, DICLOFENAC SODIUM ER, DICLOFENAC SODIUM MICRONIZED, DICLOFENAC SODIUM-MISOPROSTOL, DIFLUNISAL, DOLOBID, EC-NAPROSYN, ELYXYB, ETODOLAC, ETODOLAC ER, FELDENE, FENOPROFEN CALCIUM, FENOPRON, FLURBIPROFEN, HYDROCODONE-IBUPROFEN, IBU, IBUPAK, IBUPROFEN, IBUPROFEN LYSINE, IBUPROFEN-FAMOTIDINE, INDOCIN, INDOMETHACIN, INDOMETHACIN ER, INFLAMMACIN, INFLATHERM(DICLOFENAC-MENTHOL), KETOPROFEN, KETOPROFEN MICRONIZED, KETOROLAC TROMETHAMINE, LODINE, LOFENA, LURBIRO, MB CAPS, MECLOFENAMATE SODIUM, MEFENAMIC ACID, MELOXICAM, NABUMETONE, NABUMETONE MICRONIZED, NALFON, NAPRELAN, NAPROSYN, NAPROTIN, NAPROXEN, NAPROXEN SODIUM, NAPROXEN SODIUM CR, NAPROXEN SODIUM ER, NAPROXEN-ESOMEPRAZOLE MAG, NEOPROFEN, NORGESIC, NORGESIC FORTE, ORPHENADRINE-ASPIRIN-CAFFEINE, ORPHENGESIC FORTE, ORUDIS, OXAPROZIN, PHENYL SALICYLATE, PHENYLBUTAZONE, PIROXICAM, R.E.C.K.(ROPIV-EPI-CLON-KETOR), RELAFEN DS, ROPIVACAINE-CLONIDINE-KETOROLC, ROPIVACAINE-KETOROLAC-KETAMINE, SALSALATE, SODIUM SALICYLATE, SPRIX, SULINDAC, SUMATRIPTAN SUCC-NAPROXEN SOD, SYMBRAVO, TOLECTIN 600, TOLMETIN SODIUM, TORONOVA II SUIK, TORONOVA SUIK, TOXICOLOGY SALIVA COLLECTION, TRESNI, TREXIMET, URIMAR-T, URNEVA, VIVLODEX, VYSCOXA, XIFYRM, ZIPSOR, ZORVOLEX, ZYBIC, ZYNRELEF |
| Beta-Blockers, Oral/Rifamycins SEVERITY LEVEL: 3-Moderate Interaction: Assess the risk to the patient and take action as needed. MECHANISM OF ACTION: Rifampin is a well recognized enzyme inducer that increases the clearance of many drugs that are metabolized.(1) Beta-blockers that are extensively metabolized may be affected by rifampin. CLINICAL EFFECTS: Decreased pharmacologic effects of certain beta-blockers. PREDISPOSING FACTORS: Dose ranging of rifampin did not suggest a dose proportional interaction.(2) PATIENT MANAGEMENT: Monitor the patient's response to beta-blocker therapy when starting or stopping treatment with rifampin and adjust the dose accordingly. DISCUSSION: Controlled studies involving healthy volunteers have demonstrated rifampin to increase the clearance of metoprolol and propranolol by more than two fold.(2),(3),(4) Steady state plasma concentrations of propranolol were also reduced. The elimination half-life and protein binding of propranolol were not altered by rifampin. In a study in eight subjects, the concurrent administration of rifampin and carvedilol decreased carvedilol concentrations by 70%.(5) |
PRIFTIN, RIFABUTIN, RIFADIN, RIFAMPIN, TALICIA |
| Clonidine/Beta-Blockers SEVERITY LEVEL: 3-Moderate Interaction: Assess the risk to the patient and take action as needed. MECHANISM OF ACTION: Withdrawal of clonidine triggers increased catecholamine release. Beta-blockers inhibit the vasodilation mediated by the beta 2 receptor, leaving the vasoconstriction mediated by the alpha 2 receptor unopposed. In addition, concurrent use is expected to produce additive effects on blood pressure and heart rate requiring standard monitoring precautions. CLINICAL EFFECTS: Severe hypertension may occur upon abrupt discontinuation of clonidine in patients receiving both clonidine and beta-blockers. In addition, concurrent use is expected to produce additive effects on blood pressure and heart rate requiring standard monitoring precautions. PREDISPOSING FACTORS: None determined. PATIENT MANAGEMENT: In a patient receiving both drugs, discontinuation of the beta-blocker prior to clonidine may decrease the occurrence of rebound hypertension. If clonidine is discontinued first, rebound hypertension can be treated by restarting the clonidine or by the IV administration of phentolamine, phenoxybenzamine or prazosin. When adding either of these agents to the drug regimen of the patient, monitor blood pressure. Since labetalol has both alpha and beta activity, administration of labetalol may prevent rebound hypertension in patients undergoing clonidine withdrawal, although conflicting reports exist. In addition, concurrent use is expected to produce additive effects on blood pressure and heart rate requiring standard monitoring precautions. DISCUSSION: Increased blood pressure has been observed in patients following: 1) the discontinuation of clonidine in patients receiving beta-blockers, 2) the replacement of clonidine therapy with beta-blockers, 3) the simultaneous discontinuation of both drugs. Conflicting reports exist on the development of increased blood pressure after clonidine withdrawal in patients receiving labetalol. Patients receiving labetalol who are being withdrawn from clonidine should still be closely monitored. |
CATAPRES-TTS 1, CATAPRES-TTS 2, CATAPRES-TTS 3, CLONIDINE, CLONIDINE HCL, CLONIDINE HCL ER, DURACLON, JAVADIN, NEXICLON XR, ONYDA XR, R.E.C.K.(ROPIV-EPI-CLON-KETOR), ROPIVACAINE-CLONIDINE-KETOROLC |
| Selected Beta-Blockers/Amiodarone; Dronedarone SEVERITY LEVEL: 3-Moderate Interaction: Assess the risk to the patient and take action as needed. MECHANISM OF ACTION: The mechanism by which carvedilol, metoprolol, nebivolol, and propranolol and amiodarone produce severe bradycardia and hypotension is due to depressant effects on the sinus and AV node. Since these beta-blockers are cleared by CYP2D6 metabolism and amiodarone is a weak 2D6 inhibitor; amiodarone may decrease their metabolism.(1,3) Dronedarone is a moderate CYP2D6 inhibitor therefore may inhibit the metabolism of carvedilol, metoprolol, nebivolol and propranolol since these beta-blockers are cleared by CYP2D6 metabolism.(2-3) CLINICAL EFFECTS: The concurrent administration of hepatically metabolized beta-blockers with amiodarone(1) or dronedarone may result in bradycardia and hypotension.(1) PREDISPOSING FACTORS: None determined. PATIENT MANAGEMENT: Use low doses of beta-blockers initially. Only increase the dosage of the beta-blocker after ECG verification of good tolerability.(2) Patients receiving a concurrent therapy should be closely monitored for adverse effects, such as bradycardia and hypotension. Patients experiencing this drug interaction should have their beta-adrenergic blocking drug discontinued. Supportive therapy with sympathomimetic agents may be required. DISCUSSION: In one case report, a patient taking amiodarone developed hypotension and atropine-resistant sinus bradycardia after receiving a single dose of metoprolol. Three hours after receiving the metoprolol dose, the patient experienced dizziness, weakness, and blurred vision.(1) Another report described two patients who exhibited an interaction between amiodarone and propranolol. One patient maintained on amiodarone experienced cardiac arrest after a single oral dose of propranolol. The second patient received intravenous amiodarone followed by 2 doses of oral propranolol and developed severe bradycardia followed by ventricular fibrillation.(4) The stereoselective effect of amiodarone on the pharmacokinetics of racemic carvedilol was evaluated in 106 patients, where 52 received carvedilol monotherapy and 54 received carvedilol with amiodarone. There was no significant differences between the serum concentration to dose ratio between the 2 groups. However, there was an increase in the ratio of S-carvedilol to R-carvedilol. During amiodarone, the concentration of S-carvedilol increased from 3.03 ng/ml to 6.54 ng/ml.(5) Several studies have shown that the addition of carvedilol to congestive heart failure patients currently receiving amiodarone resulted in improved hypotension/dizziness, primary AV block, and aggravated angina.(6-8) Dronedarone increased propranolol and metoprolol (exact dosages not stated) by 1.3-fold and 1.6-fold, respectively.(2) Amiodarone increased metoprolol maximum concentration (Cmax) from 40 mcg/L to 70 mcg/L and area-under-curve (AUC) from 767 mcg x h/L to 1387 mcg x h/L after an amiodarone loading dose of 1.2 g. The interaction was noted to be more pronounced in patients with >/= 2 compared to 1 functional CYP2D6 alleles.(9) Concomitant use of dronedarone and metoprolol were studied in 49 health subjects with four differing CYP2D6 mutations. Thirty-nine were extensive metabolizers of CYP2D6 with Cmax significantly increased from baseline (134.1 ng/mL) on day 13 to 162.6 ng/mL, 195.58 ng/mL, and 180.9 ng/mL after administration of dronedarone 800 mg, 1200 mg, and 1600 mg dose, respectively.(10) |
AMIODARONE HCL, AMIODARONE HCL-D5W, MULTAQ, NEXTERONE, PACERONE |
| Nebivolol/Fluoxetine SEVERITY LEVEL: 3-Moderate Interaction: Assess the risk to the patient and take action as needed. MECHANISM OF ACTION: Fluoxetine may inhibit the metabolism of nebivolol by CYP2D6.(1) CLINICAL EFFECTS: Concurrent fluoxetine may result in elevated levels of and increased effects from nebivolol.(1) PREDISPOSING FACTORS: None determined. PATIENT MANAGEMENT: Patients receiving concurrent fluoxetine should be closely monitored and their dosage of nebivolol should be adjusted according to blood pressure response.(1) DISCUSSION: In a study in 10 healthy subjects, pretreatment with fluoxetine (20 mg daily for 21 days) increased the area-under-curve (AUC) and maximum concentration (Cmax) of a single dose of nebivolol (10 mg) by 8-fold and 3-fold, respectively.(1) In a study in 10 healthy extensive metabolizers, pretreatment with fluoxetine (20 mg daily for 21 days) increased the AUC and Cmax of a single dose of nebivolol (10 mg) by 6-fold and 2.3-fold.(3) One or more of the drug pairs linked to this monograph have been included in a list of interactions that could be considered for classification as "non-interruptive" in EHR systems. This DDI subset was vetted by an expert panel commissioned by the U.S. Office of the National Coordinator (ONC) for Health Information Technology. |
FLUOXETINE DR, FLUOXETINE HCL, OLANZAPINE-FLUOXETINE HCL, PROZAC |
| Selected CYP2D6 Substrates/Terbinafine SEVERITY LEVEL: 3-Moderate Interaction: Assess the risk to the patient and take action as needed. MECHANISM OF ACTION: Terbinafine is a strong inhibitor of CYP2D6 and may convert patients from the extensive metabolizer to poor metabolizer phenotype for this enzyme.(1) CLINICAL EFFECTS: Concurrent use of terbinafine may result in increased serum levels and adverse effects of drugs primarily metabolized by or sensitive to changes in the activity of the CYP2D6 metabolic pathway.(1,2) PREDISPOSING FACTORS: With paroxetine, the risk of anticholinergic toxicities including cognitive decline, delirium, falls and fractures is increased in geriatric patients using more than one medicine with anticholinergic properties.(3) PATIENT MANAGEMENT: Terbinafine has a serum half-life of approximately 36 hours, so the maximal effect of this interaction may be delayed for one to two weeks. Extended monitoring may be necessary. Patients receiving therapy with agents primarily metabolized by CYP2D6 need increased monitoring for adverse effects and may need a dose reduction. Plasma level monitoring should be considered in patients receiving flecainide.(4) The effect of terbinafine on CYP2D6 substrates may last for up to four weeks after terbinafine discontinuation. Over time, patients previously stabilized on the combination of terbinafine and a selected CYP2D6 substrate may need an increase in the dose of the CYP2D6 metabolized drug. DISCUSSION: In a randomized, placebo-controlled trial in 12 healthy subjects, terbinafine (150 mg daily for 6 days) increased the area-under-curve (AUC) and maximum concentration (Cmax) of a single dose of paroxetine (20 mg) by 2.9-fold and 1.9-fold, respectively.(6) In a placebo-controlled trial in 12 healthy males, terbinafine (250 mg for 4 days) increased the Cmax and AUC of a single dose of venlafaxine (75 mg) by 2.67-fold and 4.9-fold, respectively.(7) CYP2D6 substrates linked to this monograph are: dapoxetine, flecainide, metoprolol, nebivolol, paroxetine, perphenazine, propafenone, propranolol, venlafaxine and yohimbine. |
TERBINAFINE HCL |
| Selected Sensitive CYP2D6 Substrates/Mirabegron SEVERITY LEVEL: 3-Moderate Interaction: Assess the risk to the patient and take action as needed. MECHANISM OF ACTION: Mirabegron is considered a moderate inhibitor of CYP2D6. FDA defines a moderate inhibitor as a drug which increases the area-under-curve (AUC) of a sensitive substrate 2 to 5 fold.(1,2) CLINICAL EFFECTS: Concurrent use of mirabegron may lead to increased serum levels and adverse effects of drugs sensitive to inhibition of the CYP2D6 pathway.(1) Agents linked to this monograph are: atomoxetine, clomipramine, desipramine, duloxetine, imipramine, metoprolol, nebivolol, nortriptyline and yohimbine. PREDISPOSING FACTORS: With desipramine, clomipramine, imipramine and nortriptyline, the risk of seizures may be increased in patients with a history of head trauma or prior seizure; CNS tumor; severe hepatic cirrhosis; excessive use of alcohol or sedatives; addiction to opiates, cocaine, or stimulants; use of over-the-counter stimulants and anorectics; diabetics treated with oral hypoglycemics or insulin; or with concomitant medications known to lower seizure threshold (antipsychotics, theophylline, systemic steroids). The risk of anticholinergic toxicities including cognitive decline, delirium, falls and fractures is increased in geriatric patients using more than one medicine with anticholinergic properties.(3) PATIENT MANAGEMENT: Mirabegron has a long half-life of approximately 50 hours so extended monitoring over 10 to 13 days may be required to evaluate the full effect of this interaction. The manufacturer of mirabegron recommends appropriate monitoring and dose adjustment if necessary for drugs with a narrow therapeutic index.(1) Weigh the risks versus benefits of mirabegron treatment for overactive bladder symptoms based upon the interacting medication and patient specific characteristics. DISCUSSION: The manufacturer of mirabegron conducted interaction studies with two sensitive substrates of CYP2D6: desipramine and metoprolol.(1) Mirabegron 160 mg daily for 5 days increased the AUC of a single dose of metoprolol 100mg by 229%. Mirabegron 100 mg daily for 18 days increased the AUC of a single dose of desipramine by 241%. |
MIRABEGRON ER, MYRBETRIQ |
| Selected MAOIs/Selected Antihypertensive Agents SEVERITY LEVEL: 3-Moderate Interaction: Assess the risk to the patient and take action as needed. MECHANISM OF ACTION: Both MAOIs and antihypertensive agents may increase the risk of postural hypotension.(1,2) CLINICAL EFFECTS: Postural hypotension may occur with concurrent therapy of MAOIs and antihypertensive agents.(1,2) PREDISPOSING FACTORS: None determined. PATIENT MANAGEMENT: The manufacturer of phenelzine states all patients should be followed closely for symptoms of postural hypotension. Hypotensive side effects have occurred in patients who have been hypertensive and normotensive, as well as hypotensive at initiation of phenelzine.(1) The manufacturer of tranylcypromine states hypotension has been observed most commonly but not exclusively in patients with pre-existing hypertension. Tranylcypromine doses greater than 30 mg daily have a major side effect of postural hypotension and can lead to syncope. Gradual dose titration is recommended to decrease risk of postural hypotension. Combined use with other agents known to cause hypotension have shown to have additive side effects and should be monitored closely.(2) Monitor the patient for signs and symptoms of postural hypotension including dizziness, lightheadedness, or weakness, especially upon standing. Monitor blood pressure as well as orthostatic vitals and adjust antihypertensive therapy, including decreasing the dose, dividing doses, or scheduling doses at bedtime, as needed to maintain goal blood pressure. If blood pressure remains hypotensive, consider decreasing the dose of phenelzine or tranylcypromine. In some cases, discontinuation of one or both agents may be necessary.(3) Normotensive patients on stable antihypertensive therapy who are started on either phenelzine or tranylcypromine may be at increased risk for hypotension. Hypertensive patients on stable phenelzine or tranylcypromine who require antihypertensive therapy would be at decreased risk for hypotension. DISCUSSION: A review article describes the pharmacology of phenelzine and tranylcypromine as non-selective MAOIs which inhibit both type A and type B substrates. Orthostatic hypotension is described as the most common MAOI side effect and usually occurs between initiation and the first 3-4 weeks of therapy.(3) In a double-blind study, 71 patients were randomized to receive a 4-week trial of either tranylcypromine, amitriptyline, or the combination. The number of patients reporting dizziness at 4 weeks was not different between the three treatment groups (tranylcypromine 52.4%; amitriptyline 65%; combination 66.7%). Blood pressure (BP) assessment noted a significant drop in standing BP in the tranylcypromine group compared to baseline (systolic BP change = -10 mmHg; p<0.02 and diastolic BP change = -9 mmHg; p<0.02). Combination therapy also had a significant drop in standing BP compared to baseline (systolic BP change = -9 mmHg; p<0.02). Patients receiving amitriptyline had no significant change in BP from baseline at 4 weeks. All three groups had a trend toward increasing orthostatic hypotension in BP changes from lying to standing. The change in orthostatic hypotension was significant in the amitriptyline group with an average systolic BP orthostatic drop of -9 mmHg (p<0.05).(4) A randomized, double-blind study of 16 inpatients with major depressive disorder were treated with either phenelzine or tranylcypromine. Cardiovascular assessments were completed at baseline and after 6 weeks of treatment. After 6 weeks, 5/7 patients (71%) who received phenelzine had a decrease in standing systolic BP greater than 20 mmHg from baseline. Head-up tilt systolic and diastolic BP decreased from baseline in patients on phenelzine (98/61 mmHg v. 127/65 mmHg, respectively; systolic change p=0.02 and diastolic change p=0.02). After 6 weeks, 6/9 patients (67%) who received tranylcypromine had a decrease in standing systolic BP greater than 20 mmHg from baseline. Head-up tilt systolic and diastolic BP decreased from baseline in patients on tranylcypromine (113/71 mmHg v. 133/69 mmHg, respectively; systolic change p=0.09 and diastolic change p=0.07).(5) Selected MAOIs linked to this monograph include: phenelzine and tranylcypromine. Selected antihypertensive agents include: ACE inhibitors, alpha blockers, ARBs, beta blockers, calcium channel blockers, aprocitentan, clonidine, hydralazine, moxonidine, and sparsentan. |
NARDIL, PARNATE, PHENELZINE SULFATE, TRANYLCYPROMINE SULFATE |
| Tizanidine/Selected Antihypertensives SEVERITY LEVEL: 3-Moderate Interaction: Assess the risk to the patient and take action as needed. MECHANISM OF ACTION: Tizanidine is an alpha-2 agonist. Concurrent use with antihypertensive agents may result in additive effects on blood pressure.(1) CLINICAL EFFECTS: Concurrent use of antihypertensives and tizanidine may result in hypotension.(1) PREDISPOSING FACTORS: None determined. PATIENT MANAGEMENT: Patients receiving concurrent therapy should be monitored for hypotension. The risk of hypotension may be decreased by careful titration of tizanidine dosages and monitoring for hypotension prior to dose advancement. Counsel patients about the risk of orthostatic hypotension.(1) DISCUSSION: Severe hypotension has been reported following the addition of tizanidine to existing lisinopril therapy.(2-4) |
ONTRALFY, TIZANIDINE HCL, ZANAFLEX |
| Selected CYP2D6 Substrates/Desvenlafaxine SEVERITY LEVEL: 3-Moderate Interaction: Assess the risk to the patient and take action as needed. MECHANISM OF ACTION: Desvenlafaxine is considered a weak inhibitor of CYP2D6.(1) CLINICAL EFFECTS: Concurrent use of desvenlafaxine may lead to increased serum levels and adverse effects of drugs sensitive to inhibition of the CYP2D6 pathway.(1) Agents linked to this monograph are: atomoxetine, dapoxetine, deutetrabenazine, dextromethorphan, metoprolol, nebivolol, perphenazine, tolterodine, and yohimbine. PREDISPOSING FACTORS: With perphenazine and tolterodine, the risk of anticholinergic toxicities including cognitive decline, delirium, falls and fractures is increased in geriatric patients using more than one medicine with anticholinergic properties.(2) PATIENT MANAGEMENT: Reduce the dose of CYP2D6 substrates by up to one-half when coadministered with desvenlafaxine 400 mg.(1) Studies have shown that desvenlafaxine does not have a clinically relevant effect on CYP2D6 metabolism at the dose of 100 mg daily. CYP2D6 substrates should be dosed at the original level when coadministered with desvenlafaxine 100 mg or lower or when desvenlafaxine is discontinued.(1) The manufacturer of desvenlafaxine does not provide recommendations for use of desvenlafaxine at dosages between 100-400 mg concurrently with CYP2D6 substrates. Monitor for toxicity of the CYP2D6 substrate and consider reducing its dosage. DISCUSSION: In a study, coadministration of desvenlafaxine 100 mg daily with desipramine (single dose 50 mg) increased desipramine's maximum concentration (Cmax) and area-under-the-curve (AUC)by 25% and 17%.(1) In a study, coadministration of desvenlafaxine 400 mg daily with desipramine (single dose 50 mg) increased desipramine's maximum concentration (Cmax) and area-under-the-curve (AUC)by 50% and 90%.(1) Selected CYP2D6 substrates linked to this monograph are: atomoxetine, dapoxetine, deutetrabenazine, dextromethorphan, metoprolol, nebivolol, perphenazine, tolterodine, and yohimbine. |
DESVENLAFAXINE ER, DESVENLAFAXINE SUCCINATE ER, PRISTIQ |
| Lacosamide/Beta-Blockers; Diltiazem; Verapamil SEVERITY LEVEL: 3-Moderate Interaction: Assess the risk to the patient and take action as needed. MECHANISM OF ACTION: Lacosamide may enhance the slow inactivation of voltage-gated sodium channels and may cause dose-dependent bradycardia, prolongation of the PR interval, atrioventricular (AV) block, or ventricular tachyarrhythmia.(1) CLINICAL EFFECTS: Concurrent use of lacosamide and agents that affect cardiac conduction (beta-blockers, non-dihydropyridine calcium channel blockers) may increase the risk of bradycardia, prolongation of the PR interval, atrioventricular (AV) block, or ventricular tachyarrhythmia.(1) PREDISPOSING FACTORS: None determined. PATIENT MANAGEMENT: Lacosamide should be used with caution in patients on concomitant medications that affect cardiac conduction, including beta-blockers and non-dihydropyridine calcium channel blockers.(1) If concurrent use is needed, obtain an ECG before lacosamide therapy and after lacosamide dose is titrated to steady-state.(1) Patients should be monitored closely when lacosamide is given intravenously.(1) DISCUSSION: In a clinical trial in patients with partial-onset seizures, asymptomatic first-degree atrioventricular (AV) block occurred in 4/944 (0.4%) of patient who received lacosamide compared to 0/364 (0%) with placebo.(1) In a clinical trial in patients with diabetic neuropathy, asymptomatic first-degree AV block occurred in 5/1023 (0.5%) of patients who received lacosamide compared to 0/291 (0%) with placebo.(1) Second-degree and complete AV block have been reported in patients with seizures.(1) One case of profound bradycardia was observed in a patient during a 15-minute infusion of 150 mg of lacosamide.(1) Two postmarketing reports of third-degree AV block in patients with significant cardiac history and also receiving metoprolol and amlodipine during infusion of lacosamide injection at doses higher than recommended have been reported.(1) A case report of an 88 year old female taking bisoprolol documented complete AV block after initiation of lacosamide. The patient required pacemaker implementation.(2) |
LACOSAMIDE, MOTPOLY XR, VIMPAT |
| Anticholinesterases/Beta-Blockers SEVERITY LEVEL: 3-Moderate Interaction: Assess the risk to the patient and take action as needed. MECHANISM OF ACTION: Anticholinesterases inhibit plasma cholinesterases and increase cholinergic activity. Use of anticholinesterases may have vagotonic effects on heart rate (e.g. bradycardia). Concurrent use of anticholinesterases and beta-blockers may have additive effects on bradycardia.(1) CLINICAL EFFECTS: Concurrent use of anticholinesterases and beta-blockers may have additive effects on bradycardia.(1) PREDISPOSING FACTORS: None determined. PATIENT MANAGEMENT: Concurrent use of anticholinesterases and beta-blockers is not recommended. Additive effects may be increased with cardioselective beta-blockers (e.g. atenolol). Monitor patients closely if concurrent use is warranted.(1) DISCUSSION: Concurrent use of anticholinesterases and beta-blockers may have additive effects on cardiac conduction and increase the risk of bradycardia.(1) A case report of a 65 year old African American female had a witnessed a presyncopal episode followed by a true syncopal episode with concurrent use of rivastigmine and atenolol. On day 2 of the hospital stay, the patient developed bradycardia with a heart rate in the 40s and sinus pauses greater than 2 seconds. Atenolol was discontinued yet bradycardia persisted. Following discontinuation of rivastigmine, sinus pauses resolved and heart rate returned to normal.(2) A population-based cohort study in Ontario, Canada reviewed the relationship between cholinesterase inhibitor use and syncope-related outcomes over a two year period. Hospital visits for syncope were more frequent in patients receiving cholinesterase inhibitors than controls (31.5 vs 18.6 events per 1000 person-years; adjusted hazard ratio (HR) 1.76; 95% confidence interval (CI) 1.57-1.98). Other syncope-related events were also more common in patients receiving cholinesterase inhibitors than controls: hospital visits for bradycardia (6.9 vs 4.4 events per 1000 person-years; HR 1.69; 95% CI 1.32-2.15); permanent pacemaker insertion (4.7 vs 3.3 events per 1000 person-years; HR 1.49; 95% CI 1.12-2.00); and hip fracture (22.4 vs 19.8 events per 1000 person-years; HR 1.18; 95% CI 1.04-1.34).(3) A population based case-time-control study of 1,009 patients hospitalized for bradycardia within 9 months of using a cholinesterase inhibitor were reviewed for outcomes. Of these patients, 11% required pacemaker insertion during hospitalization and 4% died prior to discharge. With adjustment for temporal changes in drug utilization, hospitalization for bradycardia was associated with recent initiation of a cholinesterase inhibitor drug (adjusted odds ratio (OR) 2.13; 95% CI 1.29-3.51). Risk was similar in patients with pre-existing cardiac disease (adjusted OR 2.25; 95% CI 1.18-4.28) and those receiving negative chronotropic drugs (adjusted OR 2.34; 95% CI 1.16-4.71).(4) |
ANTICHOLIUM, BLOXIVERZ, DEMECARIUM BROMIDE, EDROPHONIUM CHLORIDE, EXELON, MESTINON, NEOSTIGMINE METHYLSULFATE, NEOSTIGMINE-STERILE WATER, PREVDUO, PYRIDOSTIGMINE BROMIDE, PYRIDOSTIGMINE BROMIDE ER, REGONOL, RIVASTIGMINE |
| Apomorphine/Selected Antihypertensives and Vasodilators SEVERITY LEVEL: 3-Moderate Interaction: Assess the risk to the patient and take action as needed. MECHANISM OF ACTION: Apomorphine causes dose-dependent decreases in blood pressure. Concurrent use with antihypertensive agents may result in additive effects on blood pressure.(1) CLINICAL EFFECTS: Concurrent use of antihypertensives and apomorphine may result in orthostatic hypotension with or without dizziness, nausea, or syncope.(1) PREDISPOSING FACTORS: The risk of orthostatic hypotension may be increased during dose escalation of apomorphine and in patients with renal or hepatic impairment.(1) PATIENT MANAGEMENT: Patients receiving concurrent therapy should be monitored for hypotension. Counsel patients about the risk of orthostatic hypotension.(1) DISCUSSION: Healthy volunteers who took sublingual nitroglycerin (0.4 mg) concomitantly with apomorphine experienced a mean largest decrease in supine systolic blood pressure (SBP) of 9.7 mm Hg and in supine diastolic blood pressure (DBP) of 9.3 mm Hg, and a mean largest decrease in standing SBP and DBP of 14.3 mm Hg and 13.5 mm Hg, respectively. The maximum decrease in SBP and DBP was 65 mm Hg and 43 mm Hg, respectively. When apomorphine was taken alone, the mean largest decrease in supine SBP and DBP was 6.1 mm Hg and 7.3 mm Hg, respectively, and in standing SBP and DBP was 6.7 mm Hg and 8.4 mm Hg, respectively.(1) |
APOKYN, APOMORPHINE HCL, ONAPGO |
| Donepezil; Galantamine/Beta-Blockers SEVERITY LEVEL: 3-Moderate Interaction: Assess the risk to the patient and take action as needed. MECHANISM OF ACTION: Anticholinesterases like donepezil and galantamine inhibit plasma cholinesterases and increase cholinergic activity. Use of anticholinesterases may have vagotonic effects on heart rate (e.g. bradycardia). Concurrent use of anticholinesterases and beta-blockers may have additive effects on bradycardia.(1,2) CLINICAL EFFECTS: Concurrent use of donepezil or galantamine with beta-blockers may have additive effects on bradycardia.(1,2) PREDISPOSING FACTORS: None determined. PATIENT MANAGEMENT: Concurrent use of anticholinesterases like donepezil or galantamine with beta-blockers is not recommended. Additive effects may be increased with cardioselective beta-blockers (e.g. atenolol). Monitor patients closely if concurrent use is warranted.(1,2) DISCUSSION: Concurrent use of anticholinesterases and beta-blockers may have additive effects on cardiac conduction and increase the risk of bradycardia.(1,2) A case report of a 65 year old African American female had a witnessed a presyncopal episode followed by a true syncopal episode with concurrent use of rivastigmine and atenolol. On day 2 of the hospital stay, the patient developed bradycardia with a heart rate in the 40s and sinus pauses greater than 2 seconds. Atenolol was discontinued yet bradycardia persisted. Following discontinuation of rivastigmine, sinus pauses resolved and heart rate returned to normal.(3) A population-based cohort study in Ontario, Canada reviewed the relationship between cholinesterase inhibitor use and syncope-related outcomes over a two year period. Hospital visits for syncope were more frequent in patients receiving cholinesterase inhibitors than controls (31.5 vs 18.6 events per 1000 person-years; adjusted hazard ratio (HR) 1.76; 95% confidence interval (CI) 1.57-1.98). Other syncope-related events were also more common in patients receiving cholinesterase inhibitors than controls: hospital visits for bradycardia (6.9 vs 4.4 events per 1000 person-years; HR 1.69; 95% CI 1.32-2.15); permanent pacemaker insertion (4.7 vs 3.3 events per 1000 person-years; HR 1.49; 95% CI 1.12-2.00); and hip fracture (22.4 vs 19.8 events per 1000 person-years; HR 1.18; 95% CI 1.04-1.34).(4) A population based case-time-control study of 1,009 patients hospitalized for bradycardia within 9 months of using a cholinesterase inhibitor were reviewed for outcomes. Of these patients, 11% required pacemaker insertion during hospitalization and 4% died prior to discharge. With adjustment for temporal changes in drug utilization, hospitalization for bradycardia was associated with recent initiation of a cholinesterase inhibitor drug (adjusted odds ratio (OR) 2.13; 95% CI 1.29-3.51). Risk was similar in patients with pre-existing cardiac disease (adjusted OR 2.25; 95% CI 1.18-4.28) and those receiving negative chronotropic drugs (adjusted OR 2.34; 95% CI 1.16-4.71).(5) |
ADLARITY, ARICEPT, DONEPEZIL HCL, DONEPEZIL HCL ODT, GALANTAMINE ER, GALANTAMINE HBR, GALANTAMINE HYDROBROMIDE, MEMANTINE HCL-DONEPEZIL HCL ER, NAMZARIC, ZUNVEYL |
| Epinephrine/Cardioselective Beta-Blockers SEVERITY LEVEL: 3-Moderate Interaction: Assess the risk to the patient and take action as needed. MECHANISM OF ACTION: Concurrent use of beta-blockers also block the beta effects of epinephrine, which results in predomination of alpha effects. The plasma clearance of epinephrine is decreased. CLINICAL EFFECTS: Concurrent use of epinephrine with beta-blockers may result in hypertension with reflex bradycardia. Epinephrine resistance in patients with anaphylaxis has been reported. PREDISPOSING FACTORS: None determined. PATIENT MANAGEMENT: Hypertension and bradycardia are less likely to occur with cardioselective beta-blockers. If both drugs are administered, monitor blood pressure carefully. Use caution when treating anaphylaxis with epinephrine since response may be poor. DISCUSSION: A 29-year-old male undergoing elective nasal septoplasty developed severe hypertension with a blood pressure of 207/123 mmHg after topical epinephrine (1:1000) was applied to the nasal mucosa. Intravenous metoprolol was administered but the patient went into cardiogenic shock thought to be a result of unopposed alpha stimulation by the combination of epinephrine and metoprolol.(1) A study observed the differences in cardiovascular responses to subcutaneous epinephrine (given to provide hemostasis during scalp incision for craniotomy) between patients who received propranolol vs. metoprolol vs. no pretreatment. While metoprolol prevented the cardiovascular effects of epinephrine infiltration, propranolol pretreatment was associated with a highly significant increase (P less than 0.01) in mean arterial pressure and a significant decrease (P less than 0.05) in heart rate.(2) A double-blind cross-over trial studied the effects of epinephrine infusion during treatment with propranolol vs. metoprolol in 8 hypertensive patients. Patients on propranolol experienced significant increases in blood pressure and systemic vascular resistance (SVR), whereas patients on metoprolol had less increase in systolic blood pressure while the diastolic pressure remained unchanged and SVR decreased.(3) In spontaneously hypertensive rats, epinephrine in combination with pindolol induced remarkable hemodynamic changes (in particular, increase in diastolic blood pressure), which were prevented by phentolamine pretreatment, whereas epinephrine with acebutolol pretreatment induced no significant hemodynamic changes.(4) |
ADRENALIN, ARTICADENT DENTAL, ARTICAINE-EPINEPHRINE, ARTICAINE-EPINEPHRINE BIT, BUFFERED LIDOCAINE-EPINEPHRINE, BUPIVACAINE HCL-EPINEPHRINE, BUPIVACAINE-DEXAMETH-EPINEPHRN, CITANEST FORTE DENTAL, EPINEPHRINE, EPINEPHRINE BITARTRATE, EPINEPHRINE HCL-0.9% NACL, EPINEPHRINE HCL-D5W, EPINEPHRINE-0.9% NACL, EPINEPHRINE-D5W, LIDOCAINE HCL-EPINEPHRINE, LIDOCAINE HCL-EPINEPHRINE-NACL, LIDOCAINE-EPINEPHRINE, LIGNOSPAN STANDARD, MARCAINE-EPINEPHRINE, ORABLOC, R.E.C.K.(ROPIV-EPI-CLON-KETOR), RACEPINEPHRINE HCL, SENSORCAINE-EPINEPHRINE, SENSORCAINE-MPF EPINEPHRINE, SEPTOCAINE, VIVACAINE, XYLOCAINE DENTAL-EPINEPHRINE, XYLOCAINE WITH EPINEPHRINE, XYLOCAINE-MPF WITH EPINEPHRINE |
| Patiromer/Bisoprolol; Carvedilol; Nebivolol SEVERITY LEVEL: 3-Moderate Interaction: Assess the risk to the patient and take action as needed. MECHANISM OF ACTION: Patiromer may bind to bisoprolol, carvedilol, and nebivolol.(1) CLINICAL EFFECTS: Concurrent use may result in decreased gastrointestinal absorption and loss of efficacy of bisoprolol, carvedilol, and nebivolol.(1) PREDISPOSING FACTORS: None determined. PATIENT MANAGEMENT: The US manufacturer of patiromer recommends administering patiromer at least 3 hours before or 3 hours after bisoprolol, carvedilol, or nebivolol.(1) DISCUSSION: An in vitro binding study found potentially clinically significant binding of bisoprolol, carvedilol, and nebivolol by patiromer. It is recommended to take these drugs 3 hours apart.(1) |
VELTASSA |
The following contraindication information is available for NEBIVOLOL HCL (nebivolol hcl):
Drug contraindication overview.
Severe bradycardia, heart block greater than first degree, cardiogenic shock, decompensated cardiac failure, sick sinus syndrome (unless a permanent pacemaker is in place), or severe hepatic impairment (Child-Pugh class C). Known hypersensitivity to nebivolol or any ingredient in the formulation.
Severe bradycardia, heart block greater than first degree, cardiogenic shock, decompensated cardiac failure, sick sinus syndrome (unless a permanent pacemaker is in place), or severe hepatic impairment (Child-Pugh class C). Known hypersensitivity to nebivolol or any ingredient in the formulation.
There are 4 contraindications.
Absolute contraindication.
| Contraindication List |
|---|
| Acute decompensated heart failure |
| Cardiogenic shock |
| Complete atrioventricular block |
| Second degree atrioventricular heart block |
There are 8 severe contraindications.
Adequate patient monitoring is recommended for safer drug use.
| Severe List |
|---|
| Bradycardia |
| Bronchospastic pulmonary disease |
| Chronic kidney disease stage 4 (severe) GFR 15-29 ml/min |
| Chronic kidney disease stage 5 (failure) GFr<15 ml/min |
| Disease of liver |
| Hypotension |
| Pregnancy |
| Sick sinus syndrome |
There are 4 moderate contraindications.
Clinically significant contraindication, where the condition can be managed or treated before the drug may be given safely.
| Moderate List |
|---|
| Diabetes mellitus |
| Hypoglycemic disorder |
| Kidney disease with likely reduction in glomerular filtration rate (GFr) |
| Peripheral vascular disease |
The following adverse reaction information is available for NEBIVOLOL HCL (nebivolol hcl):
Adverse reaction overview.
Adverse effects reported in 1% or more of patients receiving nebivolol in clinical trials and more frequently with nebivolol than with placebo include headache, fatigue, dizziness, diarrhea, nausea, insomnia, chest pain, bradycardia, dyspnea, rash, and peripheral edema. Other adverse effects reported in 1% or more of patients receiving nebivolol during worldwide clinical trials include asthenia, abdominal pain, hypercholesterolemia, hyperuricemia, and paresthesia.
Adverse effects reported in 1% or more of patients receiving nebivolol in clinical trials and more frequently with nebivolol than with placebo include headache, fatigue, dizziness, diarrhea, nausea, insomnia, chest pain, bradycardia, dyspnea, rash, and peripheral edema. Other adverse effects reported in 1% or more of patients receiving nebivolol during worldwide clinical trials include asthenia, abdominal pain, hypercholesterolemia, hyperuricemia, and paresthesia.
There are 20 severe adverse reactions.
| More Frequent | Less Frequent |
|---|---|
| None. |
Bradycardia Chest pain Dyspnea |
| Rare/Very Rare |
|---|
|
Abnormal hepatic function tests Acute myocardial infarction Acute renal failure Angioedema Asthma exacerbation Atrioventricular block Bronchospastic pulmonary disease Hypersensitivity angiitis Hypotension Mesenteric artery thrombosis Peripheral ischemia Peripheral vasoconstriction Psoriasis Pulmonary edema Syncope Thrombocytopenic disorder Urticaria |
There are 18 less severe adverse reactions.
| More Frequent | Less Frequent |
|---|---|
|
Fatigue Headache disorder |
Diarrhea Dizziness Insomnia Nausea Peripheral edema Skin rash |
| Rare/Very Rare |
|---|
|
Acute abdominal pain Drowsy Erectile dysfunction General weakness Hypercholesterolemia Paresthesia Pruritus of skin Raynaud's phenomenon Vertigo Vomiting |
The following precautions are available for NEBIVOLOL HCL (nebivolol hcl):
Safety and efficacy not established in children younger than 18 years of age.
Contraindicated
Severe Precaution
Management or Monitoring Precaution
Contraindicated
| None |
Severe Precaution
| None |
Management or Monitoring Precaution
| None |
Category C. (See Users Guide.)
Distributed into milk in rats; not known whether nebivolol is distributed into human milk. Discontinue nursing or drug.
No substantial differences in safety or efficacy relative to younger adults. (See Dosage and Administration: Special Populations.)
The following prioritized warning is available for NEBIVOLOL HCL (nebivolol hcl):
WARNING: Do not stop taking this medication without consulting your doctor. Some conditions may become worse when you suddenly stop this drug. Some people who have suddenly stopped taking similar drugs have had chest pain, heart attack, and irregular heartbeat.
If your doctor decides you should no longer use this drug, your doctor may direct you to gradually decrease your dose over 1 to 2 weeks. When gradually stopping this medication, it is recommended that you temporarily limit physical activity to decrease strain on the heart. Get medical help right away if you develop chest pain/tightness/pressure, chest pain spreading to the jaw/neck/arm, unusual sweating, trouble breathing, or fast/irregular heartbeat.
WARNING: Do not stop taking this medication without consulting your doctor. Some conditions may become worse when you suddenly stop this drug. Some people who have suddenly stopped taking similar drugs have had chest pain, heart attack, and irregular heartbeat.
If your doctor decides you should no longer use this drug, your doctor may direct you to gradually decrease your dose over 1 to 2 weeks. When gradually stopping this medication, it is recommended that you temporarily limit physical activity to decrease strain on the heart. Get medical help right away if you develop chest pain/tightness/pressure, chest pain spreading to the jaw/neck/arm, unusual sweating, trouble breathing, or fast/irregular heartbeat.
The following icd codes are available for NEBIVOLOL HCL (nebivolol hcl)'s list of indications:
| Hypertension | |
| I10 | Essential (primary) hypertension |
| I11 | Hypertensive heart disease |
| I11.0 | Hypertensive heart disease with heart failure |
| I11.9 | Hypertensive heart disease without heart failure |
| I12 | Hypertensive chronic kidney disease |
| I12.0 | Hypertensive chronic kidney disease with stage 5 chronic kidney disease or end stage renal disease |
| I12.9 | Hypertensive chronic kidney disease with stage 1 through stage 4 chronic kidney disease, or unspecified chronic kidney disease |
| I13 | Hypertensive heart and chronic kidney disease |
| I13.0 | Hypertensive heart and chronic kidney disease with heart failure and stage 1 through stage 4 chronic kidney disease, or unspecified chronic kidney disease |
| I13.1 | Hypertensive heart and chronic kidney disease without heart failure |
| I13.10 | Hypertensive heart and chronic kidney disease without heart failure, with stage 1 through stage 4 chronic kidney disease, or unspecified chronic kidney disease |
| I13.11 | Hypertensive heart and chronic kidney disease without heart failure, with stage 5 chronic kidney disease, or end stage renal disease |
| I13.2 | Hypertensive heart and chronic kidney disease with heart failure and with stage 5 chronic kidney disease, or end stage renal disease |
| I15.1 | Hypertension secondary to other renal disorders |
| I1A.0 | Resistant hypertension |
Formulary Reference Tool